Introducción
El virus de inmunodeficiencia humana (VIH) es un retrovirus compuesto por dos cadenas de ARN monocatenario rodeadas por 15 tipos de proteínas virales. El genoma de este virus contiene nueve genes, con 9,800 pares de nucleótidos. Tres de estos genes (gag, pol y env) codifican para proteínas estructurales, dos para proteínas reguladoras (tat y rev) y cuatro para proteínas accesorias (vpu, vpr, vif y nef). Existen múltiples variantes del VIH-1 debido a la gran tasa de replicación de este y a la alta tasa de error de la transcriptasa reversa, estas se conocen como cuasiespecies minoritarias. La selección de las cuasiespecies del virus son un mecanismo de escape del control del sistema inmunológico y de generación de resistencia a agentes antirretrovirales (Engelman & Cherepanov, 2012; Lahuerta, 2009).
Con base en análisis filogenéticos de distintas áreas geográficas, el VIH se divide en tipos, grupos, subtipos, sub-subtipos, formas recombinantes circulantes (FRC) y formas recombinantes únicas (FRU) (Robertson, Sharp, McCutchan, & Hahn, 1995). Los dos tipos de VIH que existen son el VIH-1 y VIH-2. El primero (VIH-1) es el causante de la mayoría de las infecciones a nivel mundial, se divide en tres grupos: Grupo M, grupo O y grupo N (Fauci et al., 2008). Actualmente se han identificado nueve subtipos de VIH-1 (A – D, F – H, J y K) y 58 formas recombinantes circulantes (FRC). El subtipo B es predominante en América, Europa y Australia, aunque solamente causa el 10 % de las infecciones a nivel mundial; el subtipo C causa casi el 50 % de las infecciones globales, mientras que el subtipo A causa 12 % de las mismas. (Pineda-Peña et al., 2013).
Las variantes de VIH-1 presentan
una separación geográfica en todo el mundo. La información proveniente de
Centroamérica es limitada. En el 2013 se realizaron ensayos filogenéticos en
base a la secuencia pol del VIH-1, en
seis países centroamericanos; evidenciando que la introducción de VIH-1 subtipo
B en Centroamérica causa la mayoría de los casos actuales (Murillo et al.,
2013). El primer caso de VIH/SIDA detectado en Guatemala fue en el año 1984,
desde entonces hasta 2009 el Centro Nacional de Epidemiología reportó 20,488
casos. La epidemia se concentra en jóvenes de 15 a 24 años. Los departamentos
más afectados son Guatemala, Retalhuleu, Izabal, Suchitepéquez, Quetzaltenango,
Sacatepéquez y Escuintla; estos concentran más del 80 % de los casos reportados
(Silvestre, 2010). En 2016, se presentaron 2,900 nuevos casos de infecciones
por VIH y 1600 muertes relacionadas con SIDA. Ese mismo año 46,000 personas
vivían con VIH, de las cuales 36 % tenían acceso a la terapia antirretroviral.
Entre las mujeres embarazadas que viven con VIH, solamente 19 % tenían acceso
al tratamiento o profilaxis para prevenir la transmisión del virus al recién
nacido. Entre las personas que viven con VIH, aproximadamente 25 % había
suprimido la carga viral (Programa Conjunto de las Naciones Unidas sobre el
VIH/sida, 2017).
En los años 2010 y 2011 se evalúo la diversidad del VIH. Para ello, se incluyeron 145 pacientes no tratados anteriormente con antirretrovirales del Hospital Roosevelt en la Ciudad de Guatemala. Se obtuvieron las secuencias pol a través del VIH plasmático. El subtipo más prevalente fue el B, con 96.6 % y se encontró una prevalencia de formas recombinantes BF1 (2.8 %) y del subtipo C (0.7 %). Este estudio fue uno de los primeros intentos de describir la diversidad del VIH. Sin embargo, no se ha realizado ningún análisis filogenético que indique las variantes que hay de este tipo (Ávila-Ríos et al., 2011).
Debido a lo anterior se realizó un estudio retrospectivo, transversal y descriptivo cuyo objetivo fue llevar a cabo la subtipificación de las secuencias de la región pol del VIH-1 obtenidas en una clínica de atención integral durante el período del 2010 al 2015, con el fin de identificar las variantes circulantes en el grupo al cual se le brinda atención. La importancia del estudio es a nivel biológico y epidemiológico; ya que al conocer los distintos subtipos de VIH-1 presentes en las muestras analizadas, es posible conocer el comportamiento epidemiológico a nivel molecular de la pandemia, la monitorización de las conexiones entre diferentes poblaciones, ayuda al entendimiento de la transmisión viral y la patogenia del SIDA, y principalmente al desarrollo de vacunas y terapias antirretrovirales eficaces (Ríos et al., 2003; Pasquier et al., 2001). Este estudio constituye el primer estudio filogenético que se realiza en secuencias virales del gen pol obtenidas durante cinco años en una clínica de atención integral de VIH del país. Por lo tanto, los resultados pueden ser considerados para el diseño de estudios epidemiológicos de mayor escala, y para determinar la relación de las variantes circulantes de VIH-1 con la respuesta al tratamiento antirretroviral que se les brinda a los pacientes.
Materiales
y métodos
Población:
Cuatrocientos pacientes seropositivos VIH-1 con fallo virológico, obtenidas en una clínica de atención integral de VIH de la ciudad de Guatemala durante los años 2010 a 2015.
Muestra:
Cuatrocientas secuencias genéticas de la región pol del VIH-1, obtenidas de la población descrita.
Recolección de datos:
Las secuencias de la región pol del HIV-1 fueron obtenidas de muestras a las cuales se les realizó la prueba de genotipo en la clínica de atención integral de VIH, en los años 2010 – 2015. Se utilizó una hoja de captura de datos en el programa Microsoft Excel 2016.
Subtipificación de las secuencias:
Para la determinación de los distintos subtipos de HIV-1 se utilizó la herramienta REGA HIV-1 Subtyping Tool – Version 3.0 (De Oliveira et al., 2014) y las secuencias obtenidas por la prueba de genotipo en formato FASTA.
Alineamiento, modelo de evolución y análisis filogenético de secuencias:
El alineamiento se realizó con el programa MEGA 7.0.21 (Kumar et al., 2016), con un gap opening penalty de 100 y un gap extension penalty de 0.1. Para determinar el modelo de evolución se utilizó el programa jModelTest 2.1.10 (Darriba et al., 2012), y el alineamiento de las secuencias en formato FASTA, la determinación del modelo se realizó bajo el criterio de Akaike (AIC) y el criterio bayesiano (BIC). Para los análisis filogenéticos, se utilizó el programa MEGA 7.0.21, se realizó un árbol filogenético utilizando el método Neighbor Joining con 100 réplicas bootstrap y el método de máxima verosimilitud, con el mismo número de réplicas e indicando el modelo de evolución obtenido con jModelTest.
Para el alineamiento se utilizó un grupo externo (outgroup) con el fin de identificar la raíz del árbol filogenético. Se utilizó el Simian Immunodeficiency Virus (SIV, por sus siglas en inglés), el cual al igual que el VIH es un retrovirus, causante de infecciones en 45 especies de primates no humanos.
Resultados
Se analizaron 400 secuencias genéticas de la región pol del VIH-1, procedentes de una clínica de atención integral de VIH de la Ciudad de Guatemala, durante los años 2010 – 2015.
En la Figura 1 se muestran los porcentajes de los distintos subtipos de VIH-1 presentes en las muestras estudiadas de Guatemala, durante los años 2010 – 2015. Se puede observar que el subtipo con mayor frecuencia es el subtipo B (71.5 %) y el de menor frecuencia son las formas recombinantes (0.25 %).
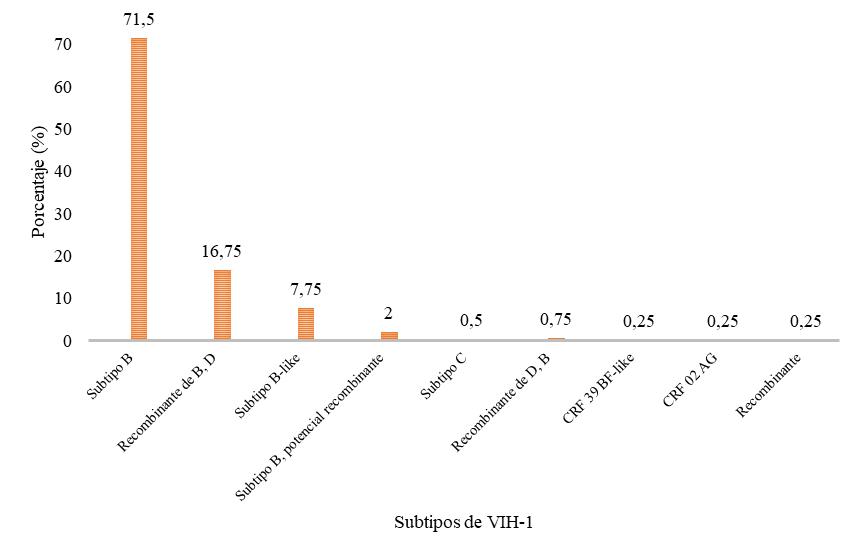 Figura
1. Porcentaje de los distintos subtipos de
VIH-1 presentes en Guatemala.
Figura
1. Porcentaje de los distintos subtipos de
VIH-1 presentes en Guatemala.
Con base en los análisis filogenéticos realizados, el modelo de evolución de VIH-1 fue general time-reversible model, con distribución gamma (GTR+G); obtenido mediante el criterio de información Akaike (AIC) y bayesiano (BIC). Este modelo incorpora distintas tasas para cada cambio y diferentes frecuencias de nucleótidos y presenta seis tipos de sustituciones. Además, el alineamiento presentó una distribución Gamma en la tasa de variación entre sitios (+G). Este modelo indica que el proceso evolutivo del VIH es completo (Arenas, 2015).
En la Figura 2, se muestra el árbol filogenético de los distintos subtipos de VIH-1 circulantes en Guatemala, realizado mediante el método de Neighbor Joining, con 100 réplicas boostrap.
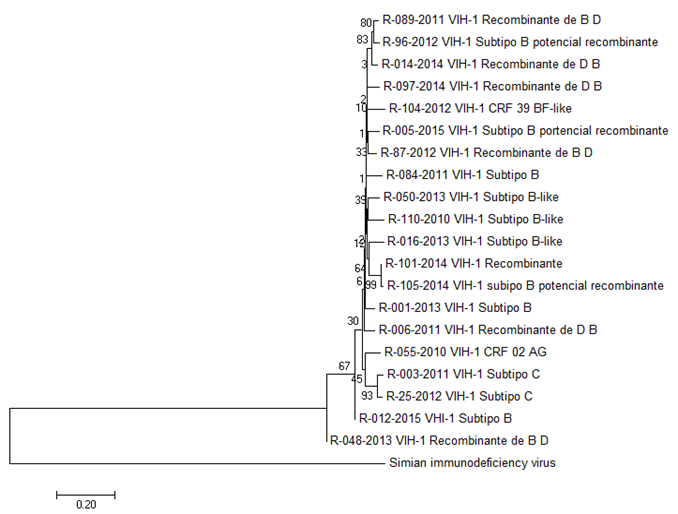 Figura
2. Árbol filogenético de los distintos
subtipos de VIH-1 circulantes en Guatemala por el método de Neighbor Joining,
realizado en MEGA 7.
Figura
2. Árbol filogenético de los distintos
subtipos de VIH-1 circulantes en Guatemala por el método de Neighbor Joining,
realizado en MEGA 7.
En la Figura 3, se observa el árbol
filogenético de los subtipos de VIH-1 circulantes en Guatemala, estimado
mediante el método de máxima verosimilitud, con 100 réplicas boostrap.
 Figura
3. Árbol filogenético de los distintos
subtipos de VIH-1 circulantes en Guatemala por el método de Máxima
Verosimilitud, realizado en MEGA 7.
Figura
3. Árbol filogenético de los distintos
subtipos de VIH-1 circulantes en Guatemala por el método de Máxima
Verosimilitud, realizado en MEGA 7.
Con relación a los datos demográficos analizados, se puede observar que el subtipo que predomina en ambos géneros, en las distintas edades y en las distintas regiones es el VIH-1 subtipo B. Los datos analizados fueron el género (Figura 4), rango de edad (Figura 5) y la procedencia de cada una de las secuencias estudiadas (Figura 6).
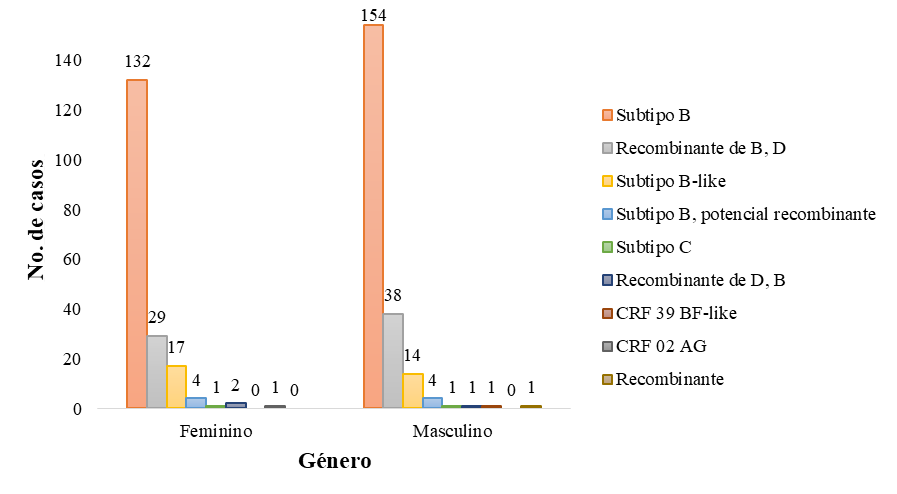 Figura
4. Subtipos de VIH-1 circulantes en
Guatemala, en base al género.
Figura
4. Subtipos de VIH-1 circulantes en
Guatemala, en base al género.
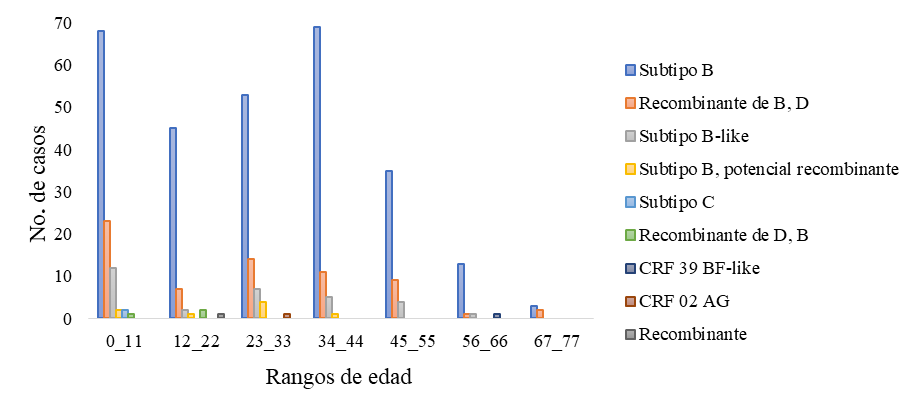 Figura
5. Subtipos de VIH-1 circulantes en
Guatemala, con base en los distintos rangos de edad de los pacientes.
Figura
5. Subtipos de VIH-1 circulantes en
Guatemala, con base en los distintos rangos de edad de los pacientes.
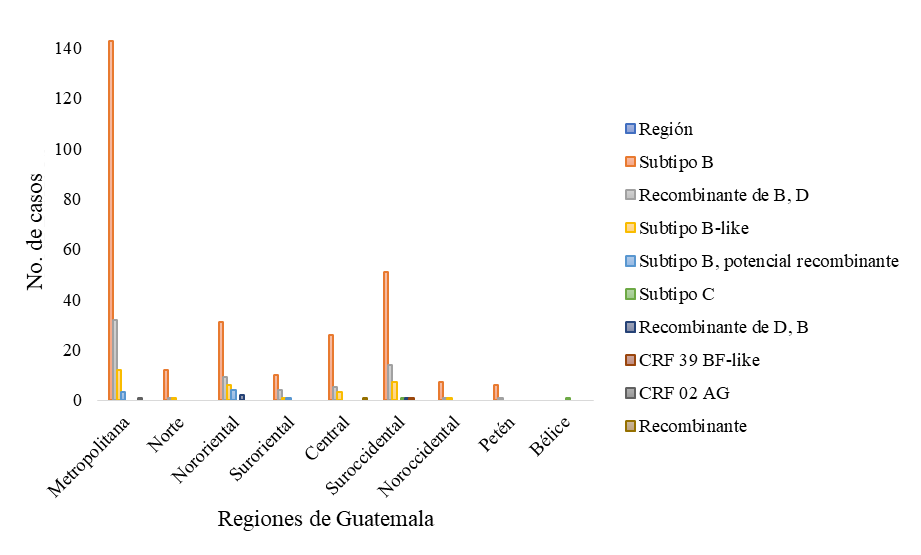 Figura
6. Subtipos de VIH-1 circulantes en
Guatemala, en base a la región procedente.
Figura
6. Subtipos de VIH-1 circulantes en
Guatemala, en base a la región procedente.
Discusión
El objetivo principal de la investigación fue llevar a cabo la subtipificación de las secuencias del gen viral pol obtenidas en una clínica de atención integral de VIH los años 2010 a 2015, para determinar las variantes circulantes del VIH-1 en Guatemala.
Como se observa en la Figura 1, el subtipo de VIH-1 con mayor prevalencia en Guatemala en pacientes VIH positivo es el VIH-1 subtipo B (71.5 %), lo cual concuerda con el estudio realizado en 2011 en el Hospital Roosevelt en la Ciudad de Guatemala, en el cual se determinó que el subtipo B es el más prevalente. El segundo subtipo más prevalente fue la forma recombinante BD (16.75 %), la cual no había sido notificada anteriormente. Se determinaron otras formas recombinantes, que se forman por recombinación de fragmentos genómicos de distintos subtipos, como CRF39_BF y CRF02 _AG. Este último es una variante no-B procedente de África y Asia; en los lugares donde predomina el subtipo B, como España, el resto de Europa y América, se ha visto una circulación creciente de subtipos no-B, como CRF02_AG, que ha sido encontrado en pacientes procedentes de África occidental (Delgado, 2011). Esto sugiere que la única secuencia de las analizadas proviene de un paciente que viajó a África o que tuvo contacto con alguna persona que estuvo en África. Por otro lado, se determinaron dos casos de subtipo C, lo que equivale al 0.5 %. Este subtipo está asociado a transmisión sexual en distintos lugares del mundo (López, Rivera-Amill, Rodríguez, Vargas, & Yamamura, 2016). La variabilidad de los subtipos de VIH-1 circulantes en Guatemala, se debe a posibles eventos de recombinación, falta de adhesión al tratamiento antirretroviral, relaciones sexuales sin protección y numerosas parejas sexuales (Taylor-Castillo et al., 2010).
Para la evaluación de las relaciones de las secuencias de los distintos subtipos de VIH-1 circulantes en Guatemala, se seleccionaron secuencias significativas de todos los subtipos identificados por REGA. Como se puede observar en las Figuras 2 y 3, en ambos árboles filogenéticos los distintos subtipos de VIH-1 se encuentra relacionados evolutivamente entre sí, ya que todos pertenecen al tipo de VIH-1, es por eso que se encuentran en un mismo clado del árbol. La principal diferencia entre ambos árboles son los valores bootstrap en cada clado, esto se debe a que con el segundo método se basa en probabilidades y la dispersión se mide en función del ajuste entre los datos observados y las predicciones calculadas por un árbol particular y el modelo de evolución. Es decir que este método es más óptimo. No todos los valores fueron mayores a 50 % en las ramas del árbol con ambos métodos, lo que indica que no todas las ramas son fiables. La razón de esto es que no se tomaron en cuenta las mutaciones por resistencia a antirretrovirales de las distintas secuencias analizadas, por lo tanto, es recomendable realizar el análisis con un alineamiento reducido para obtener valores de probabilidad más confiables.
En relación con los datos demográficos, se puede observar que el subtipo que predomina en ambos géneros, en las distintas edades y en las distintas regiones es el VIH-1 subtipo B. Con relación al subtipo C, este se detectó en un niño y una niña con un rango de edad entre 0 – 11 años, provenientes de la región suroccidente (San Marcos) y Belice. Este subtipo de VIH-1 no es muy prevalente en América, sin embargo, en el año 2011 se determinó una prevalencia de 0.7 % de este subtipo en Guatemala y es el causante de casi el 50 % de las infecciones globales (Pineda-Peña et al., 2013).
Las secuencias utilizadas son de pacientes que presentan fallo virológico, es decir que el tratamiento antirretroviral deja de reducir y mantener la carga viral de un paciente en menos de 200 copias/ml. Debido a esto, no se realizó un análisis en base al tiempo ya que no se determinaría el subtipo de VIH-1 con el cual fue infectado, el genoma de los pacientes presenta distintas mutaciones, lo que genera distintas formas recombinantes del virus. Sin embargo, se determinó menor cantidad de casos en el año 2015 en comparación al 2010, lo que significa que los pacientes son tratados adecuadamente con los distintos antirretrovirales que necesitan en base a la farmacorresistencia y farmacotoxicidad que presentan.
La variabilidad genética del VIH hace que los distintos subtipos presenten diferentes características biológicas que les confieran distinta capacidad replicativa, tropismo celular y susceptibilidad a fármacos; estos cambios genéticos pueden alterar la respuesta al tratamiento. Por lo tanto, la importancia de este estudio es a nivel biológico y epidemiológico; ya que al conocer los distintos subtipos de VIH-1 presentes en Guatemala, permite conocer el comportamiento epidemiológico a nivel molecular de la pandemia, permite la monitorización de las conexiones entre diferentes poblaciones, ayuda al entendimiento de la transmisión viral y la patogenia del SIDA, y principalmente al desarrollo de vacunas y terapias antirretrovirales eficaces (Ríos, Villanueva, San Martín, & Ramírez, 2003).
La principal limitación del estudio fue la falta de información sobre el tiempo (año) en que los pacientes fueron infectados, debido a esto no se pudo analizar la prevalencia de los distintos subtipos de VIH-1 en distintos años en Guatemala. Por lo tanto, es recomendable tomar en cuenta el año de infección y no el año de toma de muestra.
En conclusión, el subtipo de VIH-1 más predominante en Guatemala, en distintas regiones, a distintas edades y en ambos géneros es el subtipo B. Existen distintas formas recombinantes, las cuales, rpobablemente, han llegado al país por viajeros principalmente de África. Con relación a los análisis filogenéticos, podemos observar que los subtipos de VIH-1 se encuentran relacionados evolutivamente entre sí, ya que todos pertenecen al mismo tipo de VIH (VIH-1). Por lo que se encuentran en un mismo clado del árbol filogenético. Es recomendable realizar un alineamiento reducido, es decir sin tomar en cuenta las mutaciones presentes en las secuencias, con el fin de evitar valores bajos de probabilidad. Los resultados obtenidos pueden ser considerados para el diseño de estudios epidemiológicos de mayor escala en el país, para conocer las variantes del VIH-1 que circulan actualmente en Guatemala. Además, en base a los resultados se pueden realizar estudios futuros para determinar la relación de las variantes circulantes de VIH-1 con la respuesta al tratamiento antirretroviral que se les brinda a los pacientes.
Referencias
Arenas, M.
(2015). Trends in substitution models of molecular evolution. Frontiers in
Genetics, 6(319). doi:10.3389/fgene.2015.00319
Ávila-Ríos,
S., Mejía-Villatoro, C. R., García-Morales, C., Soto-Nava, M., Escobar, I.,
Mendizabal,
R., … & Reyes-Terán, G. (2011). Prevalence and patterns of VIH transmitted drug
resistance in Guatemala. Revista panamericana de salud pública, 30(6), 641-648.
Darriba,
D., Taboada, G., Doalla, R., & Posada, D. (2012). jModelTest
2: more models,
new heuristics
and parallel computing. Nature methods9(8), 772.
Delgado, R. (2011). Características
virológicas del VIH. Enfermedades
infecciosas y
microbiológica clínica, 29(1),
58-65.
De
Oliveira, T., Deforche, K., Cassol,
S., Rambaut, A., & Vandamme,
A. (2014). REGA
HIV-1 Subtyping Tool - Version 3.0 [Software]. Recuperado
de http://dbpartners.stanford.edu:8080/RegaSubtyping/stanford-hiv/typingtool/
Engelman, A., & Cherepanov, P. (2012). The structural biology of HIV-1: mechanistic
and
therapeutic insights. Nature reviews. Microbiology, 10(4), 279-290.
Fauci, A., Kasper, D., Hauser, S., Longo, D., Jameson, L.,
& Loscalzo, J. (2008). Harrison
Principios de medicina interna. México. D.F.: Mc GrawHill.
Kumar, S.,
Stecher, G., & Tamura, K. (2016). MEGA 7: Molecular Evolutionary Genetics
Analysis
Version 7.0 for Bigger Datadets. Molecular biology evolution, 33(7), 1870-4.
Lahuerta,
M. (2009). Epidemiología molecular y control de la transmisión vertical del
VIH-1:
En un área endémica de malaria del sur de Mozambique (Tesis doctoral). Universitat de Barcelona, España.
López, P.,
Rivera-Amill, V., Rodríguez, N., Vargas, F., & Yamamura, Y. (2016). The
genetic
diversity and evolution of HIV-1 subtype B epidemic in Puerto Rico. International journal of environmental research and public health, 13(1), 55.
Murillo,
W., Veras, N., Prosperi,
M., Lorenzana, I., Paz, G., Morales, S., … & Salemi, M.
(2013). A Single early introduction of VIH-1 subtype B into Central
America accounts for most current cases. Journal
of virology,87(13),7463-7470.
Pasquier, C., Millot, N., Njouom, R., Sandres, K., Cazabat, M., Puel, J., & Izopet, J. (2001).
HIV-1
subtyping using phylogenetic analysis of pol gene sequences. Journal
of virological methods, 94, 45-54.
Pineda-Peña,
A. C., Faria, N. R., Imbrechts,
S., Libin, P., Abecasis, A.
B., Deforche, K., …
& Vandamme, A. M. (2013). Automated subtyping of HIV-1 genetic sequences for
clinical and surveillance purposes: performance evaluation of the new REGA version
3 and seven other tools. Infection, genetics and evolution, 19, 337-348.
Programa Conjunto de las Naciones
Unidas sobre el VIH/sida. (2017). HIV and AIDS
Estimates. Guatemala. Recuperado de http://unaids.org/es/regionscountries/countries/guatemala
Ríos, M., Villanueva, C., San
Martín, C., & Ramírez, E. (2003). Identificación de subtipos
B y F de VIH-1 en pacientes
chilenos. Revista médica
de Chile, 131(7), 711-718.
Robertson, D., Sharp, P., McCutchan, F., &
Hahn, B. (1995). Recombination in HIV-1.
Nature,374(6518),124.
Silvestre,
L. (2010). Infecciones de transmisión
sexual en personas viviendo con VIH/SIDA
con o sin tratamiento
antirretroviral (Tesis de licenciatura). Universidad de San Carlos de
Guatemala, Guatemala.
Taylor-Castillo,
L., León-Bratti, M. P., Solano-Chinchilla, A., Herrera-Martínez, G., Boza-
Cordero,
R., León, B., … & Visoná, K. (2010). Variability in HIV-1 partial genomic sequences in Costa
Rican patients: analysis with different bioinformatics tools. Revista Panamericana
de Salud Pública, 27(1),
23-31.




