

Artículos de investigación
Algoritmo de compresión y descompresión de secuencias de ADN para su uso en traducción de proteínas
Algorithm for compressing and decompressing DNA sequences for use in protein traslation
Pädi Boletín Científico de Ciencias Básicas e Ingenierías del ICBI
Universidad Autónoma del Estado de Hidalgo, México
ISSN-e: 2007-6363
Periodicidad: Semestral
vol. 9, núm. 17, 39-45, 2021
Autor de correspondencia: afranco@uaeh.edu.mx
Resumen: La Bioinformática es una disciplina que se establece como soporte para la Biología Molecular y el estudio de genes. Es un enfoque que implementa distintas técnicas computacionales sobre datos biológicos con el objetivo de extraer información útil, para probar conocimientos existentes o incluso para crear nuevos. Sin embargo, debido a la enorme cantidad de datos y el espacio en disco, el procesamiento se vuelve complejo. Una forma de simplificar el proceso de secuencias de genes es por medio de compresión y descompresión de datos. En este artículo se propone un algoritmo que reduce el tamaño de las secuencias de ADN, sin perder información, por lo tanto reduce la complejidad de procesamiento y que además que permite la traducción parcial de la secuencia de ADN sin necesidad de descomprimir la secuencia de ADN completa.
Palabras clave: Minería de Datos, Compresión de datos, ADN, Bioinformática.
Abstract: Bioinformatics is an approach which is established as support for mulecular biology and gene estudies. This approach imple- ments several computer techniques using biological data in order to extract useful information as a proof of existing knowledge or even to create new one. Nevertheless, due to enormous amount of data and disk space, data processing becomes complex and it requires a considerable amount of resources to be processed. One way to simplify this is via DNA sequence compressing processes. This article proposes an algorithm that compresses DNA sequences, without lossing information and, in consequence, reduces time and processing and, in addition, allows partial translation of DNA sequences without the need of decompressing the whole DNA sequence.
Keywords: Data mining, Data compression, DNA, Bioinformatics.
1. Introducción
De acuerdo con Bishop(2014), se puede definir a la bioinformática como el uso de bases de datos y algoritmos de computadora para analizar protéınas, genes y la colección completa de ADN de la que está compuesto un organismo (el genoma). Para Bayat(2002), la bioinformática puede ser definida como la aplicación de tecnologías de información para almacenar, organizar y analizar una basta cantidad de datos biológicos, los cuales estan disponibles en forma de secuencias y estructuras de proteínas y acidos nucléicos. El mayor reto en biología es darle sentido a las enormes cantidades de secuencias y estructuras de datos que son generados por proyectos de secuenciadores de genomas, proteomicos y otros esfuerzos para obtener datos de biología molecular a gran escala. Las herramientas de bioinformática ayudan a revelar mecanismos fundamentales por debajo de los problemas biológicos, relacionados a la estructura y funcion de macromoléculas, rutas bioquímicas, procesos de enfermedades y evolución (Calvo, 2015). Por otro lado, todo ser vivo, también llamado organismo, comparte una serie de características o funciones, las cuales son:
Un aspecto importante a estudiar dentro de la biología es la evolución de los organismos, y es precisamente la biología evolutiva el área que establece las relaciones filogénicas, por medio del estudio de cómo se desarrollan los organismos biológicos. Además, esta disciplina permite identificar los mecanismos del desarrollo que permiten los cambios evolutivos de los individuos (Hall, 2003).
El ADN contiene información genética la cual es participe de dos procesos clave, la replicación y la expresión génica. Por medio de la replicación el ADN se duplica, permitiendo la distribución equitativa del material genético a las células hijas, como parte del proceso de división celular. Por otro lado, en la expresión génica, las enzimas leen a los genes, produciendo una síntesis de protéınas como resultado de este proceso. De manera general, se puede decir que el ADN se autorreplica, ya sea generando más ADN o transcribiéndose y generando ARN mensajero (transcripción), el cual experimenta un proceso de traducción durante la síntesis de proteínas. Francis Crick (1958) planteó que la información genética fluye del ADN al ARN y luego a las proteínas, nunca en sentido inverso, lo que se conoce como el dogma central de la biología molecular. La Figura 1 muestra este dogma, en donde se pueden observar los procesos que realizan los ácidos nucleicos (transcripción y traducción). Dichos procesos son de significante importancia en el área de la biología y en la vida misma (Calvo, 2015).
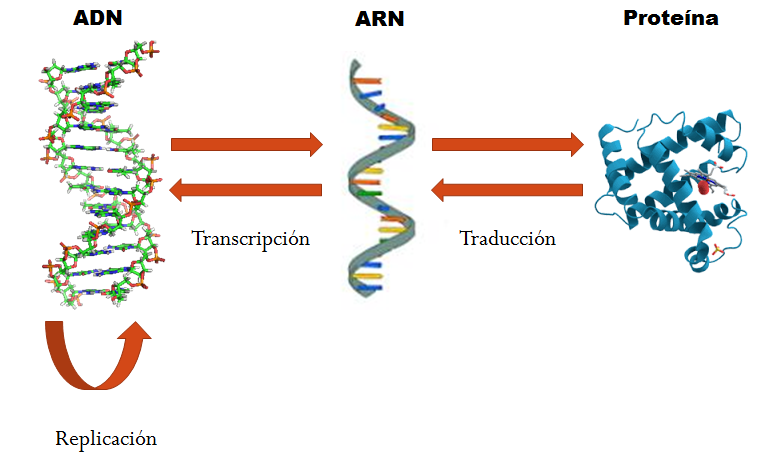
El ADN es una molécula muy grande, pero compuesta sólo por cuatro acidos nucleicos diferentes: Adenina (A), CitosinaC), Guanina (G) y Timina (T). Estos ácidos nucleicos se pueden dividir en purinas (Adenina y Guanina) y pirimidinas (Citosina y Timina). Una secuencia está formada por unidades llamadas nucleótidos. El ADN sirve como base para el proceso de transcripción, en donde se sintetiza un ARN usando como molde al ADN. En esta transcripción el ARN, a diferencia del ADN que tiene una forma de espiral de doble hélice, tiene una sola cadena de nucleótidos formada por 4 elementos: Adenina (A), Citosina (C), Guanina (G) y Uracilo (U).
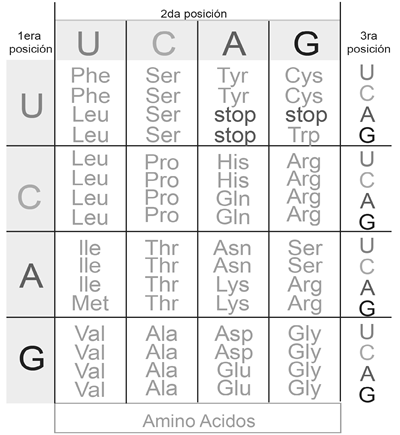
Posterior a este proceso de transcripción, a través del código genético, el ARN hace una traducción de ARN obtenido para obtener enzimas, las cuales están representadas por proteínas o aminoácidos. EL ARN mensajero se traduce haciendo uso del código genético, el cual está representado por tripletas de nucleótidos (también llamados trímeros o codones), los cuales producen 64 diferentes combinaciones de los 4 nucleótidos de ARN (por ejemplo, ACG,CGU, etc.). La Figura 2 muestra la tabla de tripletas que se usan en la traducción de ARN a protéinas. En ella se puede apreciar como se hace uso de los trímeros o codones para encontrar la relación con su aminoácido o protéına correspondiente. Ademas, la tabla 1 muestra el listado de las 22 proteínas existentes en el código genético, de los cuales el Metionina (MET - representado por el trímero ”AUG”) y los codones de parada (representados por los trímeros ”UAA”, ”UAG” y ”UGA”) sirven como instrucciones para indicar a la maquinaria celular el lugar de la cadena en el que comienza y termina la traducción del ARN mensajero, respectivamente. Por ejemplo, tomando como base la tabla mostrada en la Figura 2 puede apreciarse que el ARN mensajero ”UUU” traduce al aminoácido ”Phe” que, según el listado mostrado en la Tabla 1 representa a la proteína o aminoácido ”Fenilalanina”.
| Proteína | Código | Letra |
| Alanina | Ala | A |
| Arginina | Arg | R |
| Asparragina | Asn | N |
| Aspártico | Asp | D |
| Cisteína | Cys | C |
| Glutamina | Gln | Q |
| Glutámico | Glu | E |
| Glicina | Gly | G |
| Histidina | His | H |
| Isoleucina | Ile | I |
| Leucina | Leu | L |
| Metionina (Inicio) | Met | M |
| Fenilalanina | Phe | F |
| Lisina | Lys | K |
| Prolina | Pro | P |
| Selenocisteína | Sec | U |
| Serina | Ser | S |
| Treonina | Thr | T |
| Triptófano | Trp | W |
| Tirosina | Tyr | Y |
| Valina | Val | V |
| Parada | Stop | |
Un área que se ha vuelto importante en la bioinformática es el relacionado con la compresión de secuencias de ADN, el cual tiene algunos enfoques con el objetivo de optimizar el espacio en disco y permitir la lectura y manipulación de éstas de una manera eficaz. En sus inicios, la compresión de secuencias genómicas se llevaba a cabo utilizando herramientas de propósito general, como GZIP (Deutsch et al., 1996) o LZMA (7zip, 2019), los cuales son algoritmos compresores de textos que no omiten información de éstos al comprimirlo. La llegada del algoritmo Biocompress (Grumbach and Tahi, 1994) abrió la puerta al desarrollo de algoritmos de compresión específicos para datos biológicos. Desde enfoques probabilísticos (Pratas and Pinho, 2011; Venugopal et al., 2009) hasta enfoques donde reducen las secuencias mediante la identificación de patrones frecuentes (Kryukov et al., 2019; Zahra et al., 2019; Mansouri and Yuan, 2018; Jahaan et al., 2017; Saada and Zhang, 2015; Behzadi and Le Fessant, 2005). Todos estos algoritmos se enfocan en un proceso de codificación y decodificación de la información, de modo tal que pueden recrear la secuencia de ADN original, para de este modo aplicar el proceso del dogma de la biología molecular (Calvo, 2015), sobre la secuencia de ADN en cuestión. Además, al ser algoritmos de compresión sin pérdida, implica más tiempo de procesamiento para poderse ejecutar. En este artículo se propone un algoritmo de compresión de secuencias de ADN que permite continuar con esos estudios de una manera eficiente y permite usar directamente los procesos de transcripción y traducción de acuerdo al dogma de la biología molecular, sin la necesidad de tener que descomprimir la secuencia entera para llevarlos a cabo.
2. Descripción del método
Los biólogos han descubierto los principios de la naturaleza dirigiendo experimentos con organismos vivos. Sin embargo, debido a la falta de recursos, este método se convierte en una limitante. Sin embargo, la aparición de la bioinformática ha apoyado con dicha limitante. Bioinformática es un campo académico interdisciplinario que analiza datos biológicos por medio de técnicas modernas como lo son las ciencias computacionales, usando diversos algoritmos, estrategias y estadísticas (Bishop,2014).
Por estos esfuerzos, es posible que la bioinformática se pueda aplicar en diversos campos. A menudo es posible utilizar estas técnicas para la identificación de ciertos genes y secuencias base, así como la correlación entre ellos para de este modo poder descubrir la cura para enfermedades (Gingeras et al., 1998; Wilson et al., 2018; Liang et al., 2020; Kwon et al., 2020). No obstante, aún existen muchas desventajas del uso de la bioinformática, principalmente por la cantidad de información genómica disponible, la cual es enorme y por lo tanto el tiempo para analizarla también se vuelve considerable.
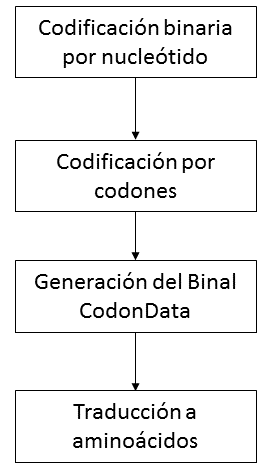
El presente trabajo propone un algoritmo de compresión de codones que, sin necesidad de llevar a cabo un proceso de descompresión de toda la secuencia de ADN, permite la traducción a protéınas ya que el resultado del algoritmo de compresión reduce la secuencia de ADN sin perder su información. La Figura 3 muestra las etapas de las que se compone dicho algoritmo.
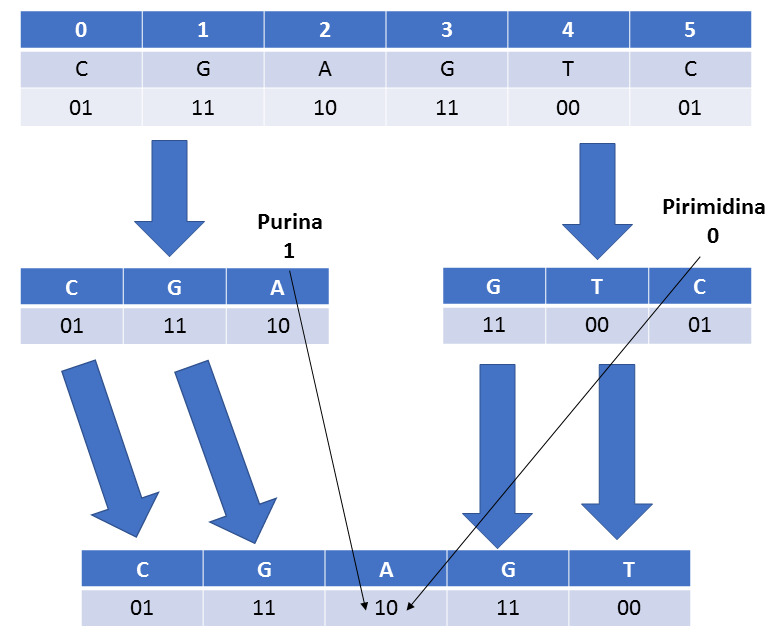
La primer etapa consiste en una codificación binaria de todos y cada uno de los nucleótidos que componen a la secuencia de ADN, utilizando como referencia la transformación mostrada en la Tabla 2. Por ejemplo, la base ”A” se convierte en ”10”, ”C” en ”01”, ”G” en 11 y por último, ”T” en 00. El primer dígito representa su clasificación (”1” - representa a las purinas y ”0” a las pirimidinas tal como se muestra en la Tabla 3), mientras que el segundo dígito representa un identificador único para cada nucleótido.
| Nucleótido | Letra | Código | Tipo |
| Adenina | A | 10 | Purina |
| Citosina | C | 01 | Pirimidina |
| Guanina | G | 11 | Purina |
| Timina | T | 00 | Pirimidina |
| Clasificación | Letras | Código |
| Purinas | A,G | 1 |
| Pirimidinas | C,T | 0 |
La segunda etapa de codificacion por codones consiste en la codificación del tercer nucleótido de cada codón. Como puede apreciarse en la Tabla 2, a excepción de casos que se verán mas adelante, el tercer nucleótido no es relevante en términos de traducción del código genético debido a que de acuerdo a su clasificación de purinas o pirimidinas se traduce a un mismo aminoácido. Esto es, en codones cuyo tercer nucleótido termina en ”A” o ”G” traducen a la misma proteína o aminoácido; de igual forma codones con terminación ”C” o ”T” (U para ARN) traducen a la misma proteína con referencia al código genético. Como se puede ver en la Figura 2, existen 64 combinaciones de tres nucleótidos, llamadas codones, que se traducen en 22 aminoácidos o proteínas. De esto se puede inferir que distintas combinaciones de ácidos nucleicos se traducen en la misma proteína. Por ejemplo: la secuencia de ARN ”UUU” y ”UUC” traducen a la proteína Fenilalanina (”phe”).
La tercera etapa consiste en la transformación de dos codones consecutivos de una secuencia de ADN en una estructura de 5 elementos llamada Codified Codon Data (CCD). Una vez creada la estructura de datos, se coloca el primer y segundo nucleótido del primer codón en la primera y segunda posición en el espacio del CCD, respectivamente. A continuación, se coloca el primer y segundo nucleótido correspondiente al segundo codón en la cuarta y quinta posición del CCD, respectivamente. El siguiente paso consiste en crear el tercer elemento del CCD. Para obtener este elemento será necesario identificar la clasificación del tercer nucleótido de cada uno de los dos codones que se toman como fuente. Utilizando la información de la Tabla 3 se codifica cada uno de los nucleótidos mencionados para generar un código de 2 dígitos. El código resultante, y con base en los datos de la Tabla 2, dará como resultado un nucleótido que será colocado en la posición faltante de llenar en el CCD. La Figura 4 muestra un ejemplo de cómo se estructura la información en el CCD.
En la ultima etapa, en la traducción a proteína, se toman los 5 elementos del CCD y se pueden expandir a 6 nucleótidos únicamente repitiendo el tercer nucleotido del CCD y colocándolo en el tercer nucleótido del segundo codón. Por ejemplo, para el CCD ”CGAGT” se puede colocar el nucleótido ”A” en la ultima posición quedando como resultado la secuencia decodificada ’CGAGTA’ para la secuencia de ADN ”CGAGTC”. Como puede observarse, a pesar de que en el segundo codón para la secuencia original de ADN (”GTC”) no es igual al segundo codón de la secuencia decodificada (”GTA”), al hacer la transcripción de la secuencia decodificada (”CGAGUC”), da como resultado la misma proteína tanto en la secuencia original como en la secuencia decodificada. De este modo, es posible realizar la traducción de dichos codones aprovechando las coincidencias de combinaciones que existen en el código genético para obtener su correspondiente secuencia de aminoácidos o proteínas sin una pérdida de información.
| Codón | Proteína o aminoácido |
| TGA | STOP |
| TGG | TPR |
| ATA | ILE |
| ATG | Met |
| Letra | Código |
| W | 02 |
| X | 12 |
| Y | 20 |
| Z | 21 |
| # | 22 |
Como se mencionó anteriormente, existen algunas excepciones de la traducción a proteínas o aminoácidos con el principio de purinas y pirimidinas. En estas excepciones, el tercer nucleótido del codón, a pesar de tener la misma clasificación, traducen a proteínas o aminoácidos diferentes, tal como lo muestra la Tabla 4. Para estos casos se agregaron codificaciones y letras adicionales que funcionan como nucleótidos, los cuales se muestran también en la Tabla 5.
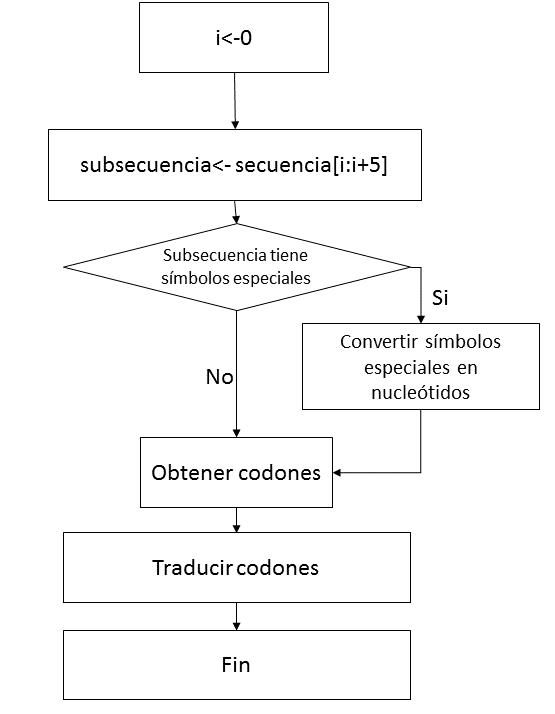
Estos cinco símbolos se utilizarán en la etapa de codificación de codones. Cuando los primeros dos nucleótidos del codón correspondan con las combinaciones ”TG” o ”AT” entonces se hará uso de estos símbolos para determinar las combinaciones que se forman de ellos y que completan el esquema para codificación. La Figura 5 muestra el proceso de obtención de los dos codones a partir del CCD.
Finalmente, el producto obtenido del proceso dará como resultado una reducción de 6 aminoácidos base a 5, de modo tal que si se codifican 24 bases el resultado se reducirá a 20, y así sucesivamente.
La Figura 6 muestra el proceso para codificar una secuencia de ADN completa. Como puede verse, el proceso recorre toda la secuencia de ADN en grupos de 6 nucleótidos, hasta el último grupo. En el momento que detecte que del final de la secuencia de ADN ya no puede extraer secuencias de dos codones o 6 nucleótidos, ese fragmento de la secuencia pasará íntegro a la codificación.
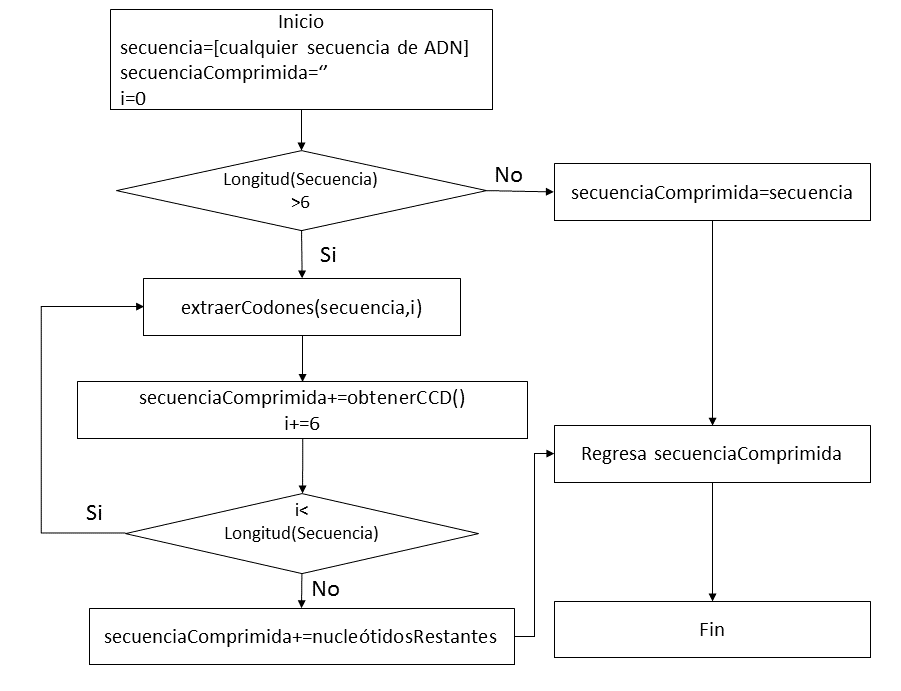
3. Resultados obtenidos
Para el presente trabajo se utilizaron 723 secuencias de ADN obtenidas de la base de datos biológica del GenBank (National Center of Biotechnology Information, 2017) con longitudes que varían desde los 185 hasta los 14,420 nucleótidos. Los experimentos se desarrollaron en lenguaje Python 3.0 en un equipo de cómputo con las siguientes características:
Procesador: AMD-A12, 3.4 GHz, 2 MB cache
Memoria RAM: 16GB DDR-4
Disco duro: 1TB
La Tabla 6 muestra el procesamiento para la secuencia de ADN ”KX358623” la cual fue procesada cada 2 codones y muestra cómo fue la codificación y decodificación para cada subsecuencia y como, al realizar los procesos de transcripción y traducción a proteínas o aminoácidos, conserva sus valores intactos, sin pérdida alguna de información.
| Subsecuencia | Proteina secuencia | CCD | Proteina CCD |
| TGGATT | WI | TGYAT | WI |
| CGAGGA | RG | CGGGG | RG |
| GATGGG | DG | GACGG | DG |
| GAACTC | EL | GAACT | EL |
| GCTGGG | AG | GCCGG | AG |
| AAGACT | KT | AAAAC | KT |
| TGTCCT | CP | TGTCC | CP |
| GATGAG | DE | GACGA | DE |
| TCGCAT | SH | TCACA | SH |
| GATGTA | DV | GACGT | DV |
| TGACAC | *H | TGACA | *H |
| CTGGGA | LG | CTGGG | LG |
| CACCCG | HP | CACCC | HP |
| CATCAG | HQ | CACCA | HQ |
Por otro lado, la Tabla 7 muestra algunos resultados para las secuencias de ADN evaluadas. Al realizar la validación de integridad de las 723 secuencias evaluadas y mencionadas previamente, se obtiene que el método genera una compresión mínima de 16.66 %, máxima de 18.91 % y media de 16.99 %, además de probar que la traducción a proteínas no tiene modificación alguna, y por lo tanto, en la secuencia de aminoácidos tampoco. La variación se debe a que, como se muestra en la Tabla 7, la secuencia no tiene longitud 6n donde n representa el número de pares de codones existentes en la secuencia. Además, en la Figura 7 y como resultado de la evaluación realizada a todas las secuencias utilizadas se puede observar que en todas ellas la traducción a aminoácidos o proteínas fue del 100 %, es decir no hubo pérdida de información y por lo tanto, el método resultante puede ser una opción a considerar para su uso en la revisión y procesamiento de secuencias de ADN.
| Folio | Long. original | Long. codificada | Porcentaje reducción |
| KX358623 | 185 | 150 | 18.919 |
| EF127130 | 250 | 205 | 18.00 |
| KU232286 | 250 | 205 | 18.00 |
| KX496872 | 361 | 300 | 16.89 |
| DQ268051 | 481 | 400 | 16.83 |
| DQ115238 | 604 | 500 | 17.21 |
| KU724099 | 605 | 500 | 17.35 |
| KX173843 | 843 | 700 | 16.96 |
| KM078971 | 969 | 805 | 16.92 |
| KX059014 | 1010 | 845 | 16.99 |
| EF463722 | 1500 | 1250 | 16.66 |
| AF047793 | 2083 | 1735 | 16.70 |
| KX101067 | 5370 | 4475 | 16.66 |
| KF383115 | 10241 | 8530 | 16.70 |
| KX576684 | 14420 | 12015 | 16.67 |
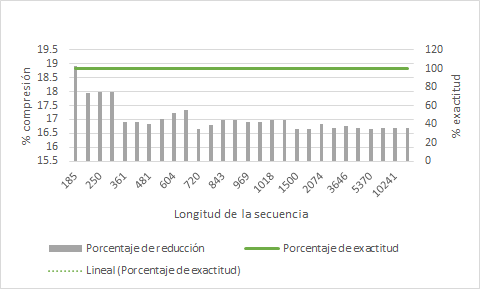
3.1. Comparación con otras metodologías
Una forma de comparar el método propuesto con otros enfoques, es a través de aspectos relacionados con sus características. La Tabla 8 muestra las características más relevantes del algoritmo propuesto en comparación con otras metodologías desarrolladas. Como puede apreciarse, mientras los algoritmos GZIP y LZMA son de propósito general, el algoritmo Biocompress y los enfoques probabilísticos, basados en identificación de patrones y el algoritmo propuesto son diseñados especialmente para comprimir secuencias de ADN. Además, a diferencia de los enfoques mencionados, el algoritmo propuesto al estar comprimido, su codificación sigue conteniendo los elementos de una secuencia de ADN (A,C,G,T), lo cual permite llevar a cabo el proceso de traducción a proteínas de un fragmento de la secuencia de ADN sin necesidad de decodificarla completamente.
4. Conclusión
El presente estudio permitió la compresión de secuencias de ADN mediante un algoritmo que aprovecha los principios del código genético, demostrando que de manera general dicho algoritmo podría ser aprovechado para reducir el número de datos de la secuencia de ADN y aún conservar su carácter de original al obtener la misma proteína o aminoácidos en su traducción. Se mostró que al tener codones decodificados con el CCD no necesariamente pueden coincidir del todo con su secuencia original y aún así pueden traducir a los mismos aminoácidos, fue precisamente este concepto el que se utilizó como una ventaja para poder implementar el algoritmo. Para los casos excepcionales, se implementó un proceso que pudiera codificar y decodificar dicha información, lo cual completa los casos y es lo que permite una traducción correcta en un 100 %. A pesar de no ser un gran porcentaje de compresión (reduce un nucleótido cada 6), al implementarlo en secuencias con longitud de millones de nucleótidos, este porcentaje representa una cantidad muy importante de espacio en disco y, en consecuencia, una complejidad menor, además de un tiempo de procesamiento eficaz. A diferencia de otros algoritmos, la traducción a aminoácidos se puede llevar a cabo directamente por pares de codones y no es necesaria la descompresión de toda la secuencia de ADN para poder completar dicha traducción. Para los expertos en bioinformática, este proceso puede ser de utilidad ya que la mayoría de sus estudios se realizan a nivel de proteınas ya que es menos complejo (3 veces menos) que una secuencia de ADN y con la propuesta del presente trabajo se puede llegar a dicho objetivo.
Agradecimientos
Agradecemos el apoyo del Área Académica de Biología del Instituto de Ciencias Básicas e Ingeniería de la Universidad Autónoma del Estado de Hidalgo, en especial al Dr. Julian Bueno Villegas por su orientación en los temas de biología.
Referencias
7zip (2019). Lzma sdk. consultado el 15-09-2020 desde https://www.7- zip.org/sdk.html/.
Bayat, A. (2002). Science, medicine, and the future: Bioinformatics. BMJ (Clinical research ed.), 324:1018–1022.
Behzadi, B. and Le Fessant, F. (2005). Dna compression challenge revisited: A dynamic programming approach. In Apostolico, A., Crochemore, M., and Park, K., editors, Combinatorial Pattern Matching, pages 190–200, Berlin, Heidelberg. Springer Berlin Heidelberg.
Bishop, O. T. (2014). Bioinformatics and Data Analysis in Microbiology. Cais- ter Academic Press.
Calvo, A. (2015). Biología celular biomédica + StudentConsult en español.
Crick, F. (1958). On protein synthesis. In Symposium of the Society for Expe- rimental Biology XII. New York: Academic Press.
Deutsch, P. et al. (1996). Gzip file format specification version 4.3. Technical report, RFC 1952, May.
Gingeras, T. R., Ghandour, G., Wang, E., Berno, A., Small, P. M., Drobniewski, F., Alland, D., Desmond, E., Holodniy, M., and Drenkow, J. (1998). Simul- taneous genotyping and species identification using hybridization pattern re- cognition analysis of generic mycobacterium dna arrays. Genome Research, 8(5):435–448.
Grumbach, S. and Tahi, F. (1994). A New Challenge for Compression Algo- rithms: Genetic Sequences. Information processing & management, 30.
Hall, B. K. (2003). Unlocking the black box between genotype and phenotype: Cell condensations as morphogenetic (modular) units. Biology and Philo- sophy, 18(2):219–247.
Jahaan, A., Ravi, T., and Panneer Arokiaraj, S. (2017). A comparative study and survey on existing dna compression techniques. International Journal of Advanced Research in Computer Science, 8(3).
Kryukov, K., Ueda, M. T., Nakagawa, S., and Imanishi, T. (2019). Nucleotide Archival Format (NAF) enables efficient lossless reference-free compression of DNA sequences. Bioinformatics, 35(19):3826–3828.
Kwon, P. S., Ren, S., Kwon, S.-J., Kizer, M. E., Kuo, L., Xie, M., Zhu, D., Zhou, F., Zhang, F., Kim, D., et al. (2020). Designer dna architecture offers precise and multivalent spatial pattern-recognition for viral sensing and inhibition. Nature chemistry, 12(1):26–35.
Liang, F., Quan, Y., Wu, A., Chen, Y., Xu, R., Zhu, Y., and Xiong, J. (2020). Insulin-resistance and depression cohort data mining to identify nutraceuti- cal related dna methylation biomarker for type 2 diabetes. Genes & Disea- ses.
Mansouri, D. and Yuan, X. (2018). One-bit dna compression algorithm. In Cheng, L., Leung, A. C. S., and Ozawa, S., editors, Neural Information Pro- cessing, pages 378–386, Cham. Springer International Publishing.
National Center of Biotechnology Information (2017). Gen- bank and wgs statistics. consultado el 15-08-2020 desde https://www.ncbi.nlm.nih.gov/genbank/.
Pratas, D. and Pinho, A. J. (2011). Compressing the human genome using exclusively markov models. In Rocha, M. P., Rodr´ıguez, J. M. C., Fdez- Riverola, F., and Valencia, A., editors, 5th International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2011), pages 213–220, Berlin, Heidelberg. Springer Berlin Heidelberg.
Vertical dna sequences compression algorithm based on hexadecimal representation.
Venugopal, K. R., Srinivasa, K. G., and Patnaik, L. M. (2009). Probabilistic Ap- proach for DNA Compression, pages 279–289. Springer Berlin Heidelberg, Berlin, Heidelberg.
Wilson, S. L., Leavey, K., Cox, B. J., and Robinson, W. P. (2018). Mining dna methylation alterations towards a classification of placental pathologies. Human molecular genetics, 27(1):135–146.
Zahra, S. E., Masood, K., and Asif, M. (2019). Dna compression using an inno- vative index based coding algorithm. In 2019 22nd International Multitopic Conference (INMIC), pages 1–6.
Apéndice
| GZIP | LZMA | Biocompress | Enfoques probabil´ısticos | Enfoques basados en identificacio´n de patrones | Algoritmo propuesto | |
| Algoritmos de propósito general | Si | Si | No | No | No | No |
| Diseñado para secuencias de ADN | No | No | Si | Si | Si | Si |
| El resultado de la codificación del algoritmo es una cadena inteligible | Si | Si | Si | Si | Si | No |
| Permite traducir parte de la secuencia de ADN sin decodificar | No | No | No | No | No | Si |
| Debe decodificar toda la secuencia de ADN para poder traducirla | Si | Si | Si | Si | Si | No |
Notas de autor
afranco@uaeh.edu.mx

