

Artículos de revisión
Revisión bibliográfica de los avances más recientes en la comprensión etiológica de la enfermedad de Alzheimer
Bibliographical review of the most recent advances in the etiological understanding of Alzheimer's disease
 Uziel Amit Téllez Ramírez uzielamit9@hotmail.com
Ana Paulina Reyes Ruiz ana00ruiz@gmail.com
Oscar Uriel Díaz Ricoy leiru.bandicoot@gmail.com
Uziel Amit Téllez Ramírez uzielamit9@hotmail.com
Ana Paulina Reyes Ruiz ana00ruiz@gmail.com
Oscar Uriel Díaz Ricoy leiru.bandicoot@gmail.com
Lux Médica
Universidad Autónoma de Aguascalientes, México
ISSN: 2007-1655
Periodicidad: Cuatrimestral
vol. 16, núm. 48, 2021
Recepción: 06 Diciembre 2020
Aprobación: 01 Junio 2021

Resumen: La enfermedad de Alzheimer (EA) es una condición neurodegenerativa caracterizada por una desintegración continua y homogénea, que ocasiona una pérdida progresiva de la memoria y deterioro de las capacidades cognitivas. La EA representa más del 60% de los casos de demencia en adultos de 65 años o más, con una afectación predominante en las áreas frontal y temporal del cerebro. Las dos hipótesis principales con respecto al desarrollo de EA son la hiperfosforilación de la proteína tau, que en situaciones normales forma parte de los axones neuronales, siendo la encargada de la unión de microtúbulos asociados al citoesqueleto de la célula, cuya malformación conduce a ovillos neurofibrilares y muerte neuronal; y la hipótesis amiloide, que describe la acumulación de placas Aβ, generadas mediante la agregación extracelular de péptidos β-amiloides insolubles. En el presente artículo de revisión se realiza una búsqueda en la literatura moderna sobre la etiología de la EA, creando una recopilación de los mecanismos más estudiados recientemente en la enfermedad, y cómo estos pueden interactuar entre sí, incluyendo los últimos avances respecto a la proteína tau, el péptido β-amiloide (Aβ), el gen APOE-4, la disfunción mitocondrial, enfermedades vasculares y la proteína alfa sinucleína, entre otras así como las principales áreas cerebrales afectadas por este padecimiento. También se evalúan distintos tratamientos empleados a partir de la etiología conocida, buscando la eliminación de placas Aβ del parénquima cerebral y la detención de la formación de los enredos neurofibrilares característicos de esta patología.
Palabras clave: enfermedad de Alzheimer, péptido β-amiloide, hiperfosforilación de TAU, Enfermedades vasculares, gen APOE-4.
Abstract: Alzheimer disease (AD) is a neurodegenerative condition, characterized by continued and homogeneous disintegration, which causes the progressive loss of memory and impairment of cognitive abilities. AD represents more than 60% of dementia in elderly people, up to 65 years old or more, with a predominant involvement of frontal and temporal brain lobes. The two main hypothesis regarding the development of AD are tau protein hyperphosphorylation, which in normal situations is part of the neural axons, as well as in charge of the union of microtubules associated to the cytoskeleton of the cell, whose malformation may result in neurofibrillary tangles and neuronal death; and amyloid hypothesis, which describes the accumulation of Aβ plaques, generated by extracellular aggregation of insoluble amyloid-β-peptide. In this review article a thorough research on modern literature rewarding Alzheimer’s disease etiology was made, creating a recompilation of the most recently studied mechanisms regarding AD, and how they may interact with each other; including the latest advances on tau protein, amyloid β-peptide, APOE-4 allele, mitochondrial dysfunction, vascular diseases and α-Synuclein protein among others; as well as the main brain regions affected by this disease. In addition, different treatments based on the known etiology were researched, looking towards the elimination of parenchymal and cerebrovascular Aβ deposits along with the cease of neurofibrillary tangles, both well-known characteristics of Alzheimer.
Keywords: Alzheimer disease, amyloid β-peptide, tau protein hyperphosphorylation, vascular diseases, APOE-4 allele.
INTRODUCCIÓN
La demencia es un síndrome clínico crónico o progresivo que afecta a distintas áreas corticales. Se caracteriza por una disminución de las capacidades intelectuales mayor que el deterioro normal por envejecimiento. Las funciones corticales afectadas incluyen la memoria, el pensamiento, la orientación, la comprensión, el cálculo, la capacidad de aprendizaje, el lenguaje y el juicio.1
La enfermedad de Alzheimer (EA) es una condición neurodegenerativa progresiva que representa más del 60% de los casos de demencia en adultos de 65 años o más.2,3,4
En 1984, el Instituto Nacional de Trastornos Neurológicos y Comunicativos y Accidentes Cerebrovasculares (NINCDS) y la Asociación de Enfermedades de Alzheimer y Trastornos Relacionados (ADRDA) publicaron por primera vez los criterios necesarios para diagnosticar esta enfermedad.5 La actualización más reciente de estos criterios fue publicada en 2011 por el Instituto Nacional del Envejecimiento y Asociación de Alzheimer (NIA-AA), y se denominó como “marco de investigación”, este se centra en el uso de biomarcadores en personas vivas para el diagnóstico de EA.6
El uso de resonancias magnéticas y fluidos biológicos pueden ayudar a determinar el proceso patológico de esta enfermedad. Los biomarcadores se clasifican en depósitos de amiloide β, tau patológica y neurodegeneración.7
Se han propuesto dos biomarcadores clave en la detección y diagnóstico de esta enfermedad: la cantidad total de tau regular y tau hiperfosforilado, y la relación en el fluido cerebroespinal entre amiloide ß40/ß42.4 Sin embargo, un estudio concluyó que personas de edad avanzada (>70 años) con estos biomarcadores afectados, tenían sólo un 20% de probabilidad de desarrollar deterioro cognitivo en cinco años, mientras que las personas con estos biomarcadores sin afectación tenían una probabilidad del 10%.8
Así, muy probablemente no sólo tau y Aß sean los responsables de causar EA. El cerebro afectado exhibe astrogliosis, atrofia de las células nerviosas y pérdida neuronal. Si bien el cerebro se caracteriza por la distribución extensiva de placas seniles (que se componen principalmente de un núcleo de péptido beta amiloide (Aβ) extracelular rodeado de terminaciones axonales agrandadas) y los ovillos neurofibrilares (NFT), los cuales son compuestos de filamentos helicoidales de la proteína tau hiperfosforilada, recientemente surgen evidencias de nuevas vías patológicas que pueden conducir al desarrollo de EA.9,10
La llamada hipótesis amiloide se basa en la creencia de que el péptido es la causa principal de la enfermedad de Alzheimer (EA). Sin embargo, múltiples estudios han tratado de demostrar que existen otras causas de semejante relevancia a la hipótesis amiloide.7,8,11,12 Otra razón que cuestiona la hipótesis amiloide como principal causa de Alzheimer es que todos los intentos de desarrollar medicamentos dirigidos al Aβ para tratar la EA han fracasado.1,13
La recopilación de información de los factores que se relacionan con la etiología de EA permite visualizar un panorama general en donde estos se interrelacionan, no sería posible distinguir las conexiones. Además, la comparativa entre ellos, tiene utilidad para determinar el grado de impacto con el que contribuye cada uno a la patogenia. Por ello, la revisión de la bibliografía moderna de cada factor etiológico se vuelve necesaria para generar nuevas perspectivas de la enfermedad y enfocar investigaciones futuras hacia los factores que prometen mejores resultados en la terapéutica.
OBJETIVO
Mediante una revisión bibliográfica, determinar los progresos recientes que se han hecho en la comprensión de la etiología de la EA, con apoyo de la medicina molecular y haciendo énfasis en los fracasos o éxitos farmacológicos modernos y antiguos que puedan determinar las causantes fisiopatológicas de la enfermedad.
METODOLOGÍA
Se consultaron diversos artículos científicos centrados en el tema de EA. Como resultado de una investigación preliminar se eligieron los factores etiológicos más frecuentes y actualizados en el desarrollo de la enfermedad de Alzheimer. Una vez identificados se inició la búsqueda de artículos con menos de cinco años de antigüedad, referentes a estos factores y artículos de investigación de nuevos fármacos que ponen a prueba las hipótesis etiológicas. La búsqueda se realizó en plataformas de divulgación científica certificada, para garantizar la validez de la información: Google Académico, Elsevier, PubMed, NCBI y Cochrane.
TAU
Tau es una de las proteínas asociadas a microtúbulos (MAP), que aumenta su estabilidad al anclarse a su dominio lateral y promover su ensamblaje. En las tauopatías su localización puede ser en el cuerpo neuronal o las dendritas.12,14,15
En el humano, tau es codificada por el gen de la proteína asociada a microtúbulos tau (MAPT), que comprende 16 exones.16 Dichos exones pueden producir seis isoformas diferentes de tau, mediante el empalme alternativo de los exones. 2, 3,10 Las isoformas de tau con tres (3R) o cuatro (4R) repeticiones de unión a microtúbulos se crean a partir del empalme alternativo del exón 10 (E10). Las isoformas de tau tienen entre 352 y 441 aminoácidos.11
La fosforilación es la principal modificación postraduccional de tau y determina su función. En el cerebro con EA, tau está hiperfosforilada, lo que inhibe su función de mantener el ensamblaje de microtúbulos, originando así inestabilidad citoesquelética neuronal.12
La tau anormal puede interactuar directamente con las nucleoporinas del complejo de poros nucleares NPC (Nuclear Pore Complex) afectando su morfología, interrumpiendo el transporte núcleo-citoplasmático en enfermedades neurodegenerativas como la EA.17,18
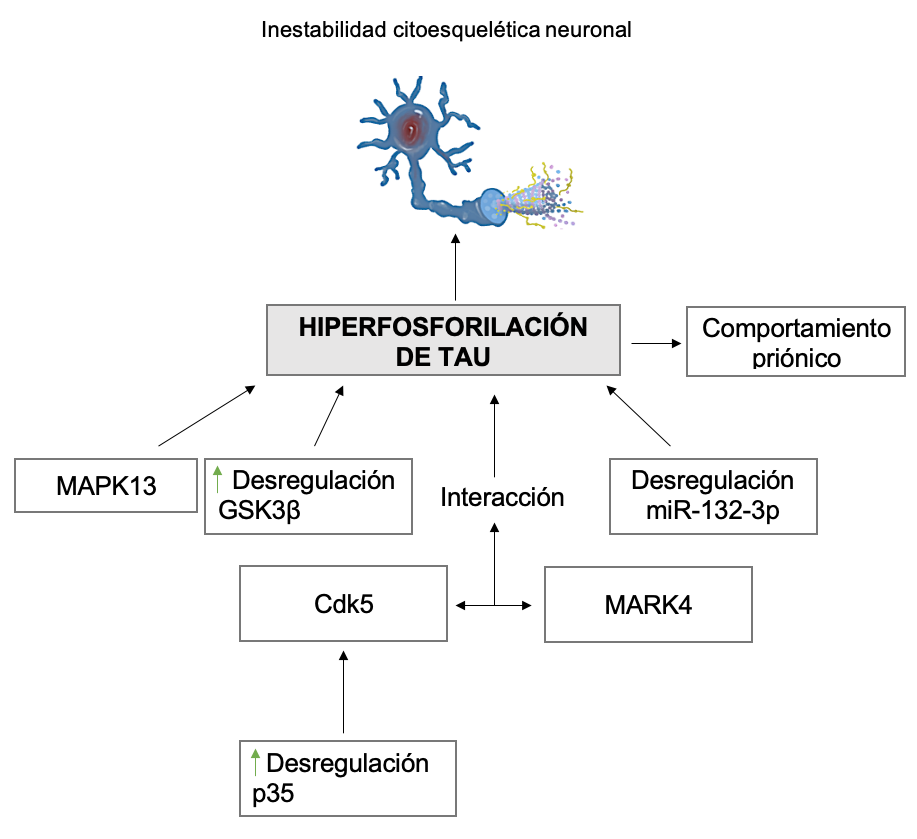
Mecanismos que pueden llevar a la hiperfosforilación de tau:
Comportamiento priónico por hiperfosforilación de tau. La tau cerebral normal contiene 2-3 moles de fosfato por mol de tau. En el cerebro patológico, esta cantidad puede ser entre tres y cuatro veces mayor. Se han identificado múltiples sitios donde tau puede hiperfosforilarse, siendo algunos residuos de Serina y Treonina especialmente susceptibles a transformar tau normal en tau patológica.19
Recientemente se inyectó la forma hiperfosforilada y oligomérica de tau en ratas sin patologías detectables. Se encontró una propiedad parecida a prión en la que se captura tau normal, ocasionando un cambio conformacional y aumentando su propagación alterando la morfología de las fibrillas proteicas, además de inducir su hiperfosforilación en sitios específicos (Ser202/Thr205, Thr212, Ser214, Thr217, Ser262, y Ser422) in vivo.19,21,22
La autoagregación fibrilar representa uno de los elementos fundamentales para los NFT.17,23 Una nueva molécula ha probado ser efectiva en ratones al reducir los niveles de tau insolubles totales y fosforilados, lo cual puede representar un importante enfoque terapeútico.24
Fosforilación de tau por distintas proteínas. La proteína quinasa activada por mitógeno 13 (MAPK13), glucógeno sintasa quinasa 3 (GSK-3) y la proteína de quinasa dependiente de ciclina 5 (Cdk5) fosforila tau in vitro.25,26 Dos subunidades de GSK (GSK3α / β) interactúan con otras 19 proteínas asociadas con la fosforilación de tau. La desregulación de GSK3β se ha implicado en la patogénesis de la EA, y la reducción de su actividad puede tener eficacia terapéutica.26 La interacción entre Cdk5 y MARK4 (quinasa afín reguladora de microtúbulos 4) fosforila tau en los sitios dirigidos de Cdk5 (especialmente Ser262). Esto resulta de la mayor activación de MARK4 ya que Cdk5 la fosforila en su dominio “spacer”.27
Hiperfosforilación de tau por distintos tipos de ARN. Existe desregulación del miR-132-3p en ratones que presentan mayor actividad de la isoforma 3R de tau. El blanco de este miARN podría estar involucrado en vías de señalización que conducen a la EA.20 También, los niveles de expresión del miR-125b son mayores en pacientes con EA y deterioro cognitivo medio (DCM).28 Por otra parte, CDK5 se encuentra regulada por la proteína p35. Cuando es desregulada, CDK5 se mueve libremente por el citoplasma fosforilando proteínas como tau.26 La p35 puede ser cortada en un fragmento C-terminal por una activación de la calpaína, generando a p10 y a p25. En la EA, la sobreactivación de la p35 y sus proteínas derivadas de fragmentaciones como la p25 pueden ser producidas por una sobreexpresión del miR-125b (Figura 1).28,29
Amiloide ß
El péptido amiloide (Aß) es una cadena de aminoácidos de longitud variable, producto de la proteolisis de la proteína precursora del amiloide (APP). Participa en diversas funciones celulares como factor neurotrófico y neuroprotector, bloqueador de apoptosis neuronal en ausencia de factores tróficos, e incluso podría atrapar iones metálicos peligrosos.30,31,32
En la EA, hay un desequilibrio entre la producción y eliminación de Aß, acumulándose en forma de placas dentro del cerebro.33
Isoformas del Aβ
Monómero Aβ. Puede considerarse una forma benigna predecesora del péptido amiloide, generada fisiológicamente por neuronas activadas en los procesos sinápticos. Los monómeros Aβ-40 y Aβ-42 contribuyen al reciclado de las vesículas sinápticas en las neuronas, cuyo proceso de liberación está sumamente controlado por la β y gamma secretasa. Especialmente, el monómero Aβ-42 posee una tendencia a agregarse, fenómeno que en magnitudes significativas en el LCR puede aumentar su viscosidad y disminuir sus niveles del monómero Aβ-42; esto último es un biomarcador temprano para pacientes con alteraciones cognitivas leves con EA y también un biomarcador parcial de la disfunción del proceso de plegamiento.34
Oligómeros de Aβ mal plegados. Provienen de cualquier monómero Aβ que mediante una transformación de su estructura tridimensional da lugar a dímeros o multímeros, adquiriendo solubilidad. Estos oligómeros se unen a proteínas y receptores.34
Fibrillas de Aβ. Isoforma de Aβ identificable en cerebros de pacientes con EA, tiene entre 39 y 43 aminoácidos, y ha sido demostrada su toxicidad en neuronas en cultivo.35 Son insolubles, formando filamentos con una estructura de lámina plegada, característica de su toxicidad. Son la principal isoforma que constituye las placas de Aβ.34
La hipótesis amiloide
El Aß ha sido propuesto como el principal precursor de la EA, causando una cascada de reacciones que conducirán finalmente a un deterioro cognitivo.36 Esto se conoce como la hipótesis amiloide. La APP es una proteína transmembrana que se puede procesar a través de dos vías mutuamente excluyentes, la amiloidogénica y la no amiloidogénica.37 En la vía amiloidogénica, la APP es procesada mediante proteólisis por la ß-secretasa o la α-secretasa. La α-secretasa produce un fragmento carboxiterminal 83 (CTF83), mientras que la ß-secretasa produce un fragmento carboxiterminal 99 (CTF99). Después, la γ-secretasa vuelve a separar los fragmentos previamente generados (CTF83 y CTF99) para formar un pequeño péptido p3 de CTF83 y Aß de CTF99. Los principales péptidos generados son Aβ40 y Aβ42.9,36,37 La proporción de Aß42 es menor con respecto al Aß40. Cuando esto se invierte, se aumenta la posibilidad de que se formen placas seniles.9
Alcances y limitaciones de la hipótesis amiloide
El gen codificante para APP se encuentra en el cromosoma 21, resultando así interesante conocer la situación de las personas con trisomía 21 con respecto a la EA, bajo la hipótesis de que estas personas tendrían 1.5 veces la cantidad normal de Aß. Los pacientes con trisomía 21 presentan esta patología alrededor de los 40 años.38 La quinurenina y el ácido quinolínico juegan un papel en el desarrollo de EA en estos pacientes. Mediante la sobreexpresión de IDO1 (enzima moduladora en la vía de la quinurenina) y la sobreexpresión de un gen estimulado por interferón (ISG), se pueden establecer dos posibles mecanismos fisiopatológicos de la EA.39,40,41
Moléculas que ayudan en la degradación de Aß
La proteína 1 relacionada con el receptor de lipoproteína (LRP1). Funciona como receptor eliminador y regula el metabolismo de los residuos peptídicos Aß llevándolos desde el líquido intersticial cerebral a la sangre sistémica; en pacientes con EA el direccionamiento de sustancias que activan al receptor LRP1 podría aumentar la eliminación del Aβ. Aún no se ha evidenciado una relación entre LRP1 y el desarrollo de EA.33,43 LRP1 también participa en la regulación del papel patogénico de ApoE.42,44
Neprilisina. La metalopeptidasa neprilisina endógena (NEP) es una enzima capaz de degradar a la proteína ß-amiloide. La administración sistémica de NEP baja los niveles de Aβ en sangre. Sin embargo, dicha administración no puede atravesar la barrera hematoencefálica.45,46
Anexina A1. La molécula de resolución anexina A1 (ANXA1) es un mediador antiinflamatorio de glucocorticoides que se asocia con procesos antiinflamatorios y ayuda con la eliminación selectiva de células similares a neuronas apoptóticas. La ANXA1 recombinante actúa limitando el proceso inflamatorio lo que resulta en una activación de la microglia, modulando la fagocitosis y la secreción de citocinas y especies neurotóxicas, ya que la ANXA1 forma una unión con el receptor 1 del péptido de formilo FPRL1. Este último sugiere un vínculo entre Aβ y FPRL1.47
Enzima degradadora de insulina (IDE). Esta enzima es capaz de degradar varios péptidos formadores de polímero Aβ. En un estudio, el tratamiento con pioglitazona en medios de cultivo de ovario de hámster transfectadas con la proteína precursora amiloide de ratón (APP) 695 redujo los niveles de Aβ 1-42 extracelular, evitando un deterioro de la memoria.48, 49
Se ha demostrado que el receptor γ activado por el proliferador de peroxisomas (PPARγ) tiene la capacidad de regular la transcripción de la IDE. PPARγ también puede unirse al elemento sensible al proliferador de peroxisomas (PPRE) en el promotor de un gen objetivo, la enzima de escisión β-amiloide 1 (BACE1).49
Activadores tisulares de plasminógenos (tPA). Son enzimas que separan al plasminógeno liberando plasmina, una proteasa capaz de degradar Aß en sus formas agregadas. El inhibidor del activador de plasminógeno (PAI-1), inhibe esta cascada de tPA/plasmina mediante su unión con tPA.50,51
En pacientes con EA la expresión de PAI-1 es elevada en zonas circundantes a las placas de Aß en el cerebro, produciendo una disminución del catabolismo del Aß.50 El uso de espinosina en ratones mejoró el déficit de actividad de la plasmina en el hipocampo.49
Enzima convertidora de angiotensina (ECA). Dentro de ECA se encontró una posible inserción (I)/ deleción (D) en el intrón 16. El gen I aumenta el riesgo de padecer EA, mientras que el gen D se asocia a la protección contra esta. ECA es capaz de degradar Aβ1-40 in vitro, generando una disminución en su agregación cerebral.53,54
Sulfato de heparán. Esta vía está mediada por la chaperona molecular clusterina, la cual captura selectivamente proteínas mal plegadas. Después sufren endocitosis a través del receptor heparán-sulfato (HS). Esta vía clusterina-HS facilita la degradación del péptido β amiloide y otras proteínas citosólicas (Figura 2).55
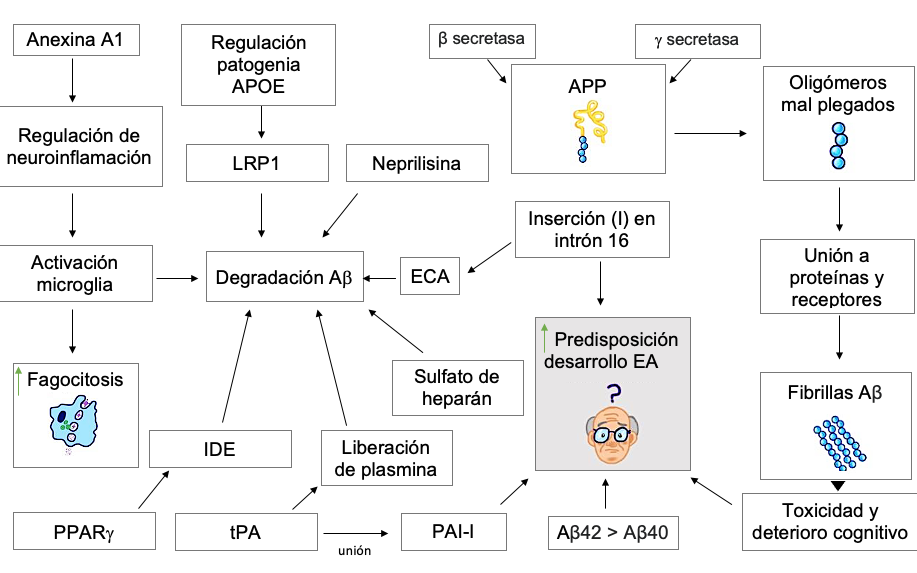
Productos finales de glicación avanzada (AGEs). Los AGEs son moléculas que se unen a los receptores para productos finales de glicación avanzada (RAGE). Esta interacción produce especies reactivas de oxígeno (ROS). En los cerebros de pacientes con EA, los niveles de los AGE son anormalmente elevados. Una posible razón es que la degradación de los AGE requiere glutatión, que se encuentra reducido en estos pacientes. Asimismo, los niveles de RAGE también son elevados en pacientes con EA. RAGE desempeña un papel crucial en la producción de Aβ y el fracaso de la degradación del Aβ. Al activarse RAGE e interaccionar con los AGE, aumentan las ROS generadas, lo que eleva la síntesis de Aß, resultando en un acúmulo de placas neurofibrilares por fallo de los mecanismos de degradación. Aunado al daño neuronal que ROS provoca, este podría ser un eje fisiopatológico de la demencia senil.56,57,58
Los fracasos farmacológicos exhiben algunos problemas de la hipótesis amiloide
Actualmente los tratamientos farmacológicos están orientados a atenuar los síntomas. Existen los inhibidores de la acetilcolinesterasa (donepezil, rivastigmina y galantamina) y aquellos con actividad antagonista del receptor de N-metil-D-aspartato no competitivo y agonista de la dopamina (memantina)59
Los anticuerpos monoclonales son fármacos orientados a curar la enfermedad, ya que reaccionan con los agregados amiloides de forma dependiente de la conformación y de la secuencia con agregados amiloides. Sin embargo, ninguno reconoce placas de Aβ en todas las condiciones.60 El bapineuzumab se sintetiza a partir de la versión para humano del anticuerpo monoclonal murino 3D6, se une a la región N-terminal del Aß y activa la fagocitosis microglial y la producción de citocinas (IL-1ß y TNFα), para que así se elimine el exceso de restos de Aß; sin embargo, con su uso no se han demostrado mejoras cognitivas, lo que exhibe una relación pobre con respecto al desarrollo de la EA.61,62 De forma similar el crenezumab reconoce especies oligoméricas y placas amiloides, con nulos efectos como tratamiento de EA.63
El solanezumab es otro fármaco de acción similar al bapineuzumab, pero se dirige al Aß soluble monomérico; no se demostraron cambios significativos en la cognición o el volumen de atrofia cerebral. El aducanumab es un fármaco que promete unirse y reducir los oligómeros solubles de amiloide, recientemente aprobado por la FDA en EUA, aunque aún existe polémica sobre su eficacia.65
Los fármacos destinados a inhibir las enzimas que separan APP (semagacestat, avagacestat, verubecestat y lanabecestat) no han mostrado ser efectivos en la reducción de la demencia moderada.66,67,68,69,70
Algunos tratamientos podrían mostrar diferentes vías fisiopatológicas que conducen a la EA
El Azeliragon es una molécula inhibidora de RAGE de administración oral. La interacción entre RAGE y amiloide resulta en daños fisiopatológicos en neuronas, microglia, astrocitos, etc.71 Sin embargo, no se ha encontrado una mejora cognitiva significativa entre los participantes con su uso.72
Por otra parte, varios miARN se han visto implicados en el desarrollo de EA. El miR-7 se ha visto involucrado en el sistema de ubiquitina proteasoma. El miR-7 se encuentra sobreexpresado en pacientes con EA, regulando negativamente la expresión de la UBE2A, una enzima conjugadora de ubiquitina. La degradación del Aß por proteólisis se ve afectada y esto podría conducir al desarrollo de placas amiloides.73 Además de este miARN, los miR34a y miR29b se han visto involucrados en al menos tres vías de degradación de Aß.74
Patogenicidad del Aβ y las placas de Aβ
Investigaciones recientes muestran que el incremento de placas de Aβ no necesariamente está relacionado con la intensificación de la EA, puesto que estas se pueden presentar tanto en individuos con daño cognitivo como en individuos cognitivamente normales. Además, ratones genéticamente modificados para la APP han presentado anormalidades fisiológicas y del comportamiento antes de la aparición de las placas de Aβ; lo cual sugiere que las placas por sí solas no son responsables de la totalidad del daño cognitivo asociado a la EA, los oligómeros de Aβ pueden desempeñar un papel más importante en la neurotoxicidad por sus efectos de sinapto toxicidad. El Aβ induce diferentes anormalidades que se presentan en la EA, como la estimulación de la formación de los NFT. Además, alteraciones neuríticas y sinápticas se presentan mayormente en zonas corticales cercanas a las placas de Aβ. En los cambios neuríticos, las dendritas de neuronas corticales pasan de ser rectas a enroscadas, se cree que tal cambio disminuye la eficiencia de transmisión de las señales nerviosas a través de las dendritas.75
La Hipótesis de la Disfunción del Beta Amiloide (BAD)
La hipótesis consiste en que el proceso fisiopatológico del Aβ comienza cuando se altera la homeostasis de la formación y metabolismo del péptido, bajo la premisa de que este desempeña un papel fisiológico. Las concentraciones del Aβ son reguladas por la actividad de la gamma y beta secretasa, y dependen de los requerimientos de cada neurona.
Los monómeros mal plegados de Aβ conducen a una deficiencia del monómero fisiológico de Aβ. La homeostasis en la concentración del monómero es esencial para la actividad sináptica. La ß-secretasa, mediante un mecanismo de retroalimentación positiva compensa, el decremento en la concentración del monómero, incrementando la producción del monómero de Aβ y la tasa de mal plegamiento. Por ende, incrementa la concentración de la forma fisiológica del monómero Aβ y también de su forma mal plegada. Comienza un ciclo vicioso en donde el mecanismo de compensación empeora la patogenicidad. Un aumento de la ß-secretasa es un marcador temprano de la EA. Además, en la EA esporádica la expresión de su gen es elevada.35,51 Esta hipótesis explica detalles que no han podido ser descritos en la teoría clásica de la cascada amiloide.
La teoría del estrés oxidativo como principal causa biológica para el desarrollo de EA
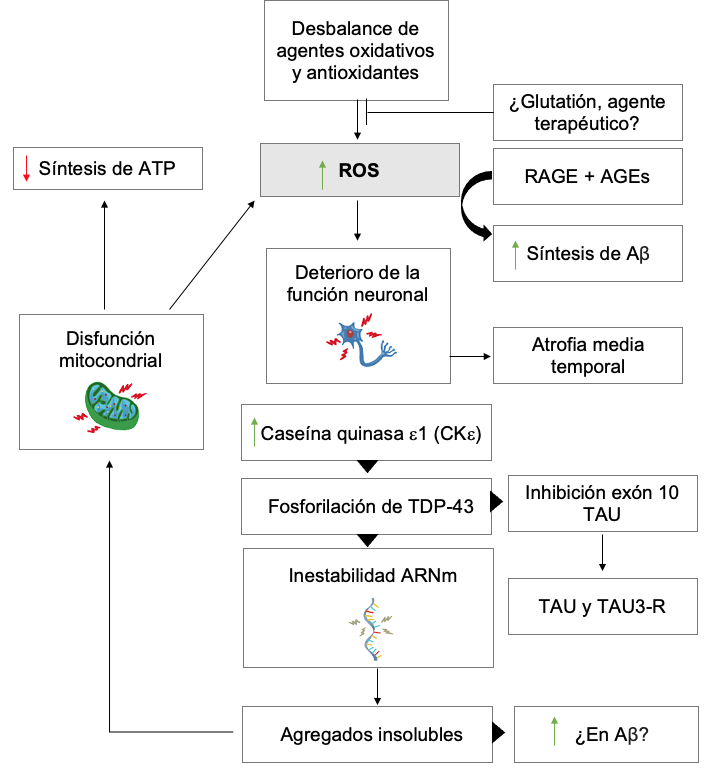
Hoy en día la hipótesis del envejecimiento más estudiada y aceptada es la teoría del estrés oxidativo, que surge cuando se produce un desbalance negativo entre los agentes oxidantes producidos y el sistema neutralizador antioxidante.76,77,78
Las ROS en exceso deterioran la función biomolecular de las células neuronales, dirigiéndose a moléculas como ADN, ARN, lípidos y proteínas. El antioxidante glutatión juega un papel importante en la prevención del estrés oxidativo, ayudando a mitigar el efecto nocivo de las ROS, por ello se ha propuesto como potencial blanco terapéutico frente a la EA.79,80,81
La guanina del ADN y ARN puede transformarse en 8-hidroxiguanina y 8-hidroxi-2-desoxiguanosina. Las proteínas pueden ser carboniladas o nitradas. Los lípidos de las membranas celulares pueden ser sometidas a peroxidación lipídica, aumentando la permeabilidad membranal; gracias a esto moléculas como el Ca+ o K+, pueden entrar libremente causando mutilación de las proteínas de membrana, enzimas y receptores, provocando desestabilización neuronal (Figura 3).79
La personalidad podría influir en el desarrollo de demencia frontotemporal y EA
Las neuronas von Economo (NvE) son un tipo de neuronas presentes en distintas áreas del cerebro, como la corteza entorrinal, la formación del hipocampo, la corteza cingulada anterior, la ínsula ventral, el área dorsomedial de Brodmann 9 (BA9) y, recientemente descubiertas, en la corteza frontopolar medial.82
Por su localización, se ha postulado que son importantes en procesos sociales superiores, atención y motivación, en el monitoreo, control de errores, y control emocional.83 En casos de demencia frontotemporal y en pacientes con EA, un estudio demostró un decremento significativo en cantidad de NvE, con respecto a pacientes sin demencia. Cabe resaltar que algunos pacientes con demencia frontotemporal no tenían tauopatías y eran positivos frente a TDP 43.84,85
TDP-43
La proteína 43 de unión al ADN TAR (TDP-43) codificada por el gen TARDBP, es una proteína de unión a ADN y ARN que bajo circunstancias fisiológicas normales se encuentra en el núcleo. Se ha visto involucrada en el empalme de los genes APOA2, CFTR y SMN. Se considera como un factor patológico secundario en la EA, su fosforilación anormal se ha visto implicada principalmente en la esclerosis lateral amiotrófica (ELA).86,87,88 La forma patológica de la TDP-43, presente en 19% a 57% de los casos de la EA esporádica, se produce por mecanismos anormales de fosforilación, ubiquitinación, y proteolisis, que la convierten en agregados insolubles, los cuales posteriormente constituyen inclusiones en los cuerpos neuronales, menos frecuentemente, en células gliales.89,90 Se observa su participación en los siguientes mecanismos subyacentes a la EA:
Interacción de TDP-43 con depósitos de Aβ. Se ha observado TDP-43 cerca de depósitos de Dominio Intracelular de APP (AICD), resultantes de la proteólisis de APP por la γ secretasa; también una relación entre la disminución de su concentración con el aumento de los niveles del Aß oligomérico y la disminución de placas de Aß, exacerbando la neurodegeneración en ratones con EA.88,90
Hiperfosforilación de tau por TDP-43. Se han observado niveles anormalmente altos de caseína quinasa ε1(CKε) en cerebros con EA, lo que puede llevar a la fosforilación de TDP-43, debido a la regulación de tau que ejerce TDP-43 mediante su ARNm, promoviendo la inestabilidad del ARNm de tau, inhibiendo además la inclusión del exón 10, elevando en consecuencia la cantidad de tau y tau-3R.91,92,93
Atrofia media temporal asociada a TDP-43. Se ha asociado mediante técnicas modernas de neuroimagen. Un estudio con 342 participantes observó un agravamiento de la memoria (53% positivos para TDP-43), y una mayor pérdida progresiva del volumen temporomedial que en los sujetos TDP negativos en todas las etapas de Braak.94
Inducción de disfunción mitocondrial por TDP-43. Se ha observado principalmente en ELA, Enfermedad de Huntington y EA.89,95 Una pequeña cantidad de la TDP-43 es transportada a la mitocondria, uniéndose al ARNm mitocondrial transcrito y regulando su traducción. En condiciones de estrés celular o mutaciones se convierte en su forma patológica fosforilada insoluble que se acumula en inclusiones mitocondriales, provocando alteraciones específicas en la transcripción y traducción del ARNm mitocondrial que afectan la morfología del organelo.89,95,96 La disfunción de este se manifiesta por la pérdida del potencial de membrana, el aumento en la producción de ROS, y la reducción de la síntesis de ATP.95,96
APOE-4
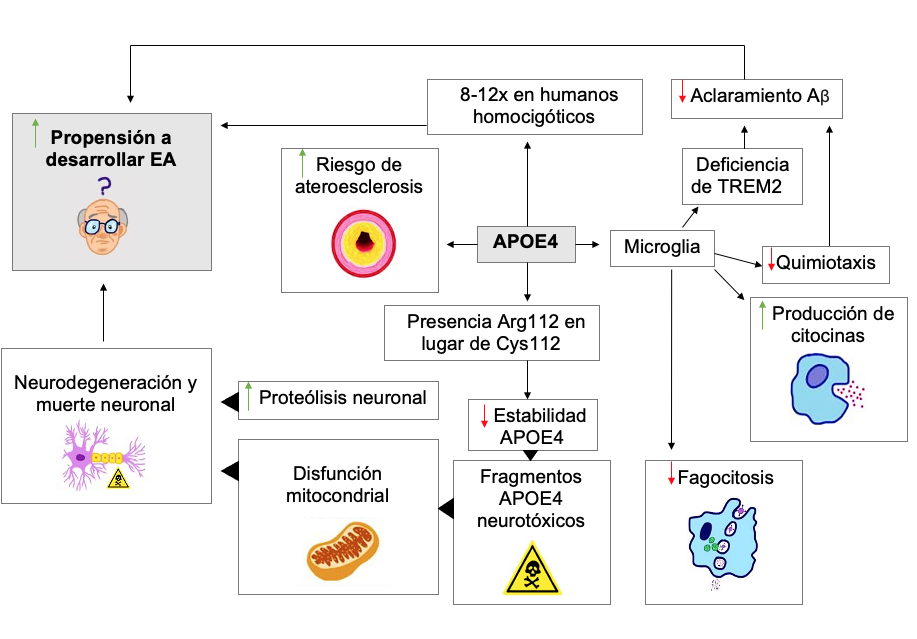
La apolipoproteína E es una glicoproteína codificada por el gen APOE, que participa en el catabolismo de lipoproteínas ricas en triglicéridos. Los tres principales alelos APOE son APOE2, APOE3 y APOE4 (ε4) que generan isoformas proteicas del mismo nombre. Dos aminoácidos y sus posiciones, específicamente 112 y 158, son la diferencia entre ellas y afectan sus funciones en el transporte de lípidos y aclaramiento de Aβ. La presencia de Arg 112 en APOE4 en lugar de Cys permite la interacción de los dominios de la proteína y reduce su estabilidad. En neuronas bajo estrés, esto exacerba la susceptibilidad a proteólisis, produciendo cambios neurodegenerativos y fragmentos de APOE4 neurotóxicos implicados en disfunción mitocondrial y muerte neuronal.100
La función celular se ve alterada en presencia de APOE4. En las células microgliales parecidas a humanas (iMGLS) disminuye su quimiocinesis a sitios lesionados y su fagocitosis, mientras aumenta la producción de citocinas en condiciones proinflamatorias.101,102 Las neuronas presentan una mayor producción de Aβ42 y mayor número de sinapsis. Los astrocitos acumulan colesterol.102
El alelo ε4 es un factor genético predisponente a padecer EA de inicio tardío, ya que del 40 al 80% de los afectados lo tienen. Sin embargo, APOE4 se asocia con un mayor riesgo de aterosclerosis y desarrollo de EA en un 8-12x en humanos homocigóticos.99 Se ha sugerido un papel de interferencia con la producción y/o degradación del Aß. Una vía propuesta se relaciona con el receptor desencadenante expresado en células mieloides 2 (TREM2). TREM2 se expresa en células microgliales, y en modelos de ratones este ayuda a la degradación de Aß al alterar su estructura. Los ratones deficientes de TREM2 exhiben mayor daño neurítico.104,105,106
La variante TREM2 R47H le confiere una pérdida de función a TREM2 regular. Esto se traduce en una expresión reducida de TREM2 alrededor de las placas y una menor afinidad de las células mieloides frente a placas Aß.104 APOE usualmente fagocita neuronas apoptóticas dependiendo de la expresión de TREM2. La variante TREM2 R47H reduce la afinidad de TREM2 por apoE.107
Por otra parte, el alelo ε2 de APOE reduce la posibilidad de desarrollar EA al disminuir la agregación del Aβ, especialmente en ausencia del alelo ε4; debido a que APOE4 suele opacar su actividad protectora cuando sí está presente, puede ser esta otra vía de patogenicidad del alelo ε4.108,109
APOE3 Christchurch (R136S) es una mutación recientemente descubierta que se cree podría proteger contra los efectos cognitivos de la EA al limitar la patología de tau.110
Los oligonucleótidos antisentido, correctores moleculares de estructuras pequeñas y las pequeñas moléculas que mejoran la isoprenilación de APOE4 mediada por transportador dependiente de ATP A1 (ABCA1) son terapias prometedoras relacionadas con APOE4.111
Los oligonucleótidos antisentido podrían reducir la expresión de APOE4, disminuyendo la cantidad de placas Aß. Estudios donde se implementó al inicio de la patogénesis en ratones sin placas de Aß han mostrado resultados positivos.112 Los otros dos métodos podrían ser efectivos para corregir y/o aminorar la formación de placas de Aß (Figura 4).113,114
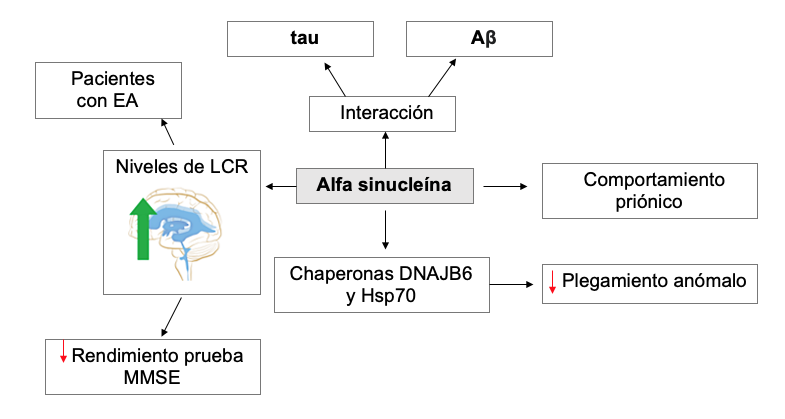
Alfa Sinucleína
Esta proteína es codificada por el gen SNCA y actúa como reguladora de los receptores de proteínas de fijación soluble de NSF (factor sensible a la N-etilmaleimida) del complejo SNARE, además de regular la apoptosis y diferenciación neuronal.115,116,117
Una investigación determinó la alfa sinucleína y beta sinucleína como dos componentes peptídicos no amiloides de las placas seniles. Se formuló la hipótesis de su acción como precursoras de las placas seniles solubles propias de enfermedades neurodegenerativas dada su tendencia a formar estructuras ß. La alfa sinucleína (140 aminoácidos) puede ser separada para formar la beta sinucleína (134 aminoácidos), por lo cual fue descrita como la precursora de los componentes no amiloides de las placas seniles.118
La alfa sinucleína es reconocida en la enfermedad de Parkinson o la demencia asociada a cuerpos de Lewy.119 Sin embargo, se han identificado concentraciones elevadas en el líquido cefalorraquídeo de pacientes con EA.120,121
La interacción electrostática entre alfa sinucleína y tau
Tanto in vitro como in vivo, estas dos proteínas se oligomerizan y agregan mutuamente. Ambas tienen un comportamiento similar a prion, así su forma patológica mal plegada puede moldear la α-sinucleína endógena, formando polímeros patológicos que se propagan en el SNC.122,123
La interacción entre alfa sinucleína y tau mal plegadas origina la formación de placas amiloides. La alfa sinucleína se puede dividir en una región N-terminal, una región central y una región C-terminal. La interacción electrostática entre la región C-terminal con carga negativa y K18 de tau con carga positiva lleva a la formación de placas amiloides (Figura 5). Las chaperonas DNAJB6 y Hsp70 reducen el plegamiento anormal de la alfa sinucleína.124,125,126
Enfermedades vasculares
La EA es la forma más común de demencia, seguida de la demencia vascular (VaD). Estudios epidemiológicos demuestran que las enfermedades vasculares son un factor de riesgo para el desarrollo de EA. Aproximadamente el 50% de los pacientes de EA presentan los principales signos de esta enfermedad, así como un historial de patologías vasculares, ambos incrementando con la edad.127, 128
Al envejecer, las arterias centrales se vuelven rígidas, generando un aumento de la tensión arterial junto con la velocidad del flujo pulsátil. El aumento en la velocidad de flujo aórtico reduce la perfusión en el lóbulo frontal y aumenta la reactividad cerebrovascular en los lóbulos frontal, temporal y occipital.129,130 Esto propone que las personas hipertensas sean más propensas a desarrollar demencia y EA en edades avanzadas. Así mismo, una hipotensión en la vida media-avanzada eleva a razón de 200% el riesgo de desarrollar EA.131,132
La hipoperfusión cerebral puede ser causada por depósitos de Aß, mediante un mecanismo que produce ROS, liberando endotelinas para la contracción de pericitos, finalizando en constricción capilar.133
Estudios demuestran un aumento de la presión significativo de 10 a 15 años antes del diagnóstico de EA, además de un posterior incremento en el acúmulo de neurofibrillas. No obstante, también se identifica una hipotensión general en los años precedentes al diagnóstico, con una disminución constante.134
Igualmente, la aterosclerosis, el acúmulo de grasa, y otras sustancias en las paredes de los vasos, son un factor común en pacientes de EA, principalmente en las arterias carótidas e iliacas; aumentando la severidad de esta con la disminución del desempeño cognitivo. Una hipótesis que relaciona la aterosclerosis y EA se puede definir de la siguiente manera:
El drenaje perivascular representa la principal vía de eliminación para el Aß, drenando del líquido intersticial al espacio perivascular y, finalmente, a los ganglios linfáticos cervicales. Análisis biomecánicos confirman que las ondas de reflexión de la pared arterial después de la onda de pulso cardiaco son las impulsoras para el tránsito del líquido intersticial. Así, al existir rigidez arterial, se reduce el movimiento de la pared del vaso, ocasionando una menor eliminación y su consecuente acumulación.135
Disfunción renal y su contribución al proceso neurodegenerativo
El daño renal, manifestado por una menor tasa estimada de filtración glomerular (TFGe), desbalance en la relación albúmina-creatinina en orina (UACR), entre otros, se relaciona con el declive cognitivo en edades avanzadas.136
Durante la enfermedad renal terminal (ERSD) hasta 87% de pacientes presenta demencia.137 El papel de los riñones como vía de eliminación del Aß se refleja en pacientes con disfunción renal crónica (CKD), quienes tienen mayores niveles de Aß en suero.138
En la disfunción renal existe relación entre la UACR y el desarrollo de EA. Una albuminuria ≥30.0 mg/g afecta a la barrera hematoencefálica y la eliminación de Aß, aumentando en un 35% el riesgo de desarrollar demencia.139,140,141 La acumulación de toxinas urémicas también contribuye al desarrollo de desórdenes neuropsiquiátricos como la EA.142Además, el infarto cerebral silencioso, lesión de sustancia blanca y atrofia cerebral son prevalentes en pacientes con daño renal severo, contribuyendo al proceso neurodegenerativo.137,141
Cascada de coagulación
Las plaquetas constituyen la fuente primaria de Aβ en el torrente sanguíneo periférico al portar elementos de la cascada amiloide, como la APP, la alfa secretasa y la beta secretasa. Los niveles plasmáticos de péptido Aβ son dependientes de la activación plaquetaria, y con esta se liberan fragmentos del péptido que se depositan en el endotelio cerebrovascular, contribuyendo al desarrollo de angiopatías.143,144También se ha sugerido que GSK3B, presente en las plaquetas, tiene una actividad incrementada en pacientes de EA, favoreciendo la fosforilación anormal de tau, con un riesgo mayor de formar ovillos neurofibrilares (Figura 6).143
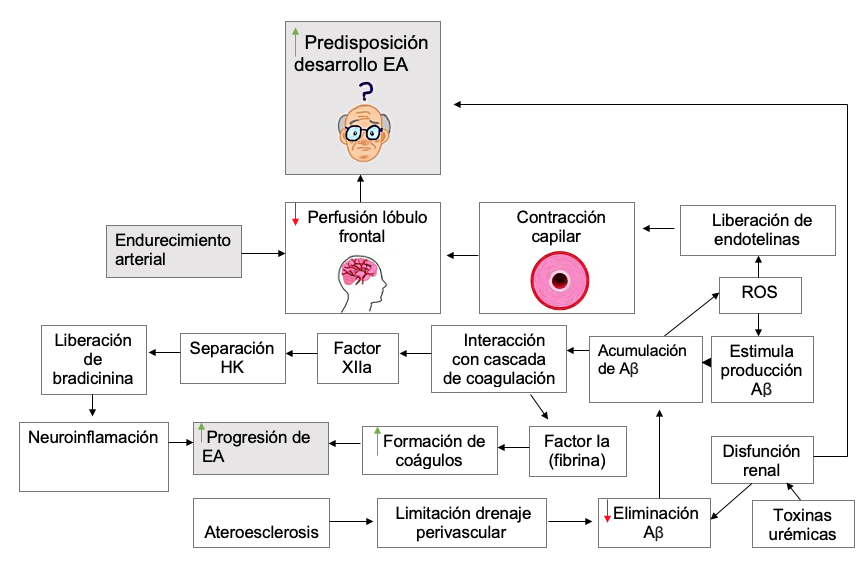
El Aß desencadena la neuroinflamación mediante el sistema de activación por contacto (CAS), el cual inicia con el factor de coagulación XII (FXII), resultando en la separación del quininógeno de alto peso molecular (HK) mediado por la calicreína plasmática liberando bradicinina proinflamatoria. El HK es un medidor de la inflamación.145 En plasma de pacientes con EA existe mayor actividad de separación de HK, así como una mayor activación de CAS en ratones inyectados con Aß42. Una depleción del FXII mejora el rendimiento cognitivo y reduce la neuroinflamación en ratones con EA.146,147 La fibrina (factor Ia) en la EA se encuentra en áreas de disfunción sináptica y patología amiloide interactuando con Aβ, lo que conlleva a una formación de coágulos. La disminución sistemática de fibrina en ratones con EA demostró mejoría en la progresión de la enfermedad, por lo que un tratamiento con anticoagulantes tradicionales resulta beneficioso para pacientes con demencia.148
DISCUSIÓN
Entre los diferentes factores etiológicos discutidos se observan puntos en común. Esta investigación presenta resultados consistentes con otros artículos en cuanto a que el alelo ε4 es considerado uno de los factores genéticos que otorgan mayor riesgo a padecer EA de inicio tardío.98,99,149 En algunos de los mecanismos evaluados se discute el rol de la edad en el avance e influencia del individuo, como en la teoría del estrés oxidativo y enfermedades vasculares. Si bien el estrés oxidativo no es exclusivo del envejecimiento, este aumenta sobre todo en personas de edades avanzadas.76 De igual manera, se ha encontrado que las enfermedades vasculares, como el endurecimiento arterial o la hipertensión, juegan un papel importante en el riesgo de desarrollo de EA, lo que coincide con otros autores.129,135
Sin lugar a duda, los avances en la medicina moderna seguirán siendo indispensables en la comprensión de patologías con etiología variada y mecanismos moleculares de difícil comprensión como lo es la EA.
CONCLUSIÓN
La enfermedad de EA es el resultado de diversas anomalías como la hiperfosforilación de tau y las placas amiloides; factores que contribuyen al desequilibrio entre la producción y la eliminación del péptido Aβ, lo cual, en ausencia de tratamiento externo, provoca la rápida evolución de la enfermedad. Cada una de las etiologías mencionadas contribuyen al desarrollo y evolución de la EA.
Gracias a los avances tecnológicos y estudios en laboratorio en animales se han planteado maneras de aumentar la eliminación directa de Aβ. Sin embargo, dado que la causa de la EA es multifactorial, no se ha podido concretar un tratamiento definitivo para los pacientes que sufren dicha enfermedad.
Referencias
1. Dementia [Internet]. World Health Organization. World Health Organization; [citado el 20 de Abril de 2020]. Disponible de: https://www.who.int/news-room/fact-sheets/detail/dementia
2. CIE-10 [Internet]. CIE-10 código F00 | Demencia en la enfermedad de Alzheimer (G30.–). [citado el 20 de Abril de 2020]. Disponible de: https://icdcode.info/espanol/cie-10/codigo-f00.html
3. What Is Alzheimer's Disease? [citado el 20 de Abril de 2020]. Disponible de: https://www.psychiatry.org/patients-families/alzheimers/what-is-alzheimers-disease
4. Hippius H, Neundörfer G. The discovery of Alzheimer's disease. Dialogues Clin Neurosci. 2003;5(1):101-8.
5. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939-44.
6. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia. 2011;7(3):263-9.
7. Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
8. Petersen RC, Lundt ES, Therneau TM, Weigand SD, Knopman DS, Mielke MM, et al. Predicting Progression to Mild Cognitive Impairment. Ann Neurol. 2019;85(1):155-60.
9. Kametani F, Hasegawa M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease. Front Neurosci. 2018 Jan30;12:
10. Kumar A. Alzheimer Disease [Internet]. StatPearls [Internet]. U.S. National Library of Medicine; 2019 [cited 2020 Mar 28]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK499922/
11. Espíndola SL, Damianich A, Alvarez RJ, Sartor M, Belforte JE, Ferrario JE, Gallo JM, Avale ME. Modulation of Tau Isoforms Imbalance Precludes Tau Pathology and Cognitive Decline in a Mouse Model of Tauopathy. Cell Rep. 2018;23(3):709-715.
12. Pîrşcoveanu DFV, Pirici I, Tudorică V, Bălşeanu TA, Albu VC, Bondari S, Bumbea AM, Pîrşcoveanu M. Tau protein in neurodegenerative diseases - a review. Rom J Morphol Embryol. 2017;58(4):1141-1150
13. dos Santos Picanco LC, Ozela PF, de Fatima de Brito Brito M, Pinheiro AA, Padilha EC, Braga FS, et al. Alzheimer's Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. CMC. 2018;25(26):3141-59.
14. Karp, G., Iwasa, J. and Marshall, W. Cell and Molecular Biology, Concepts and Experiments. 8th ed. 2019; pp.311-313.
15. Morris M, Maeda S, Vossel K, Mucke L. The Many Faces of Tau. Neuron. 2011 May;70(3):410-26.
16. Symbol report for MAPT [Internet]. HGNC. [cited 2020 Feb 29]. Available from: https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:6893
17. Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y, et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer's Disease. Neuron. 2018. 09 5;99(5):925-940.e7.
18. Floch AG, Palancade B, Doye V. Fifty years of nuclear pores and nucleocytoplasmic transport studies: multiple tools revealing complex rules. Methods Cell Biol. 2014;122:1-40.
19. Miao J, Shi R, Li L, Chen F, Zhou Y, Tung Y et al. Pathological Tau From Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS- and Reducing Agent-Resistant Aggregation of Tau in vivo. Frontiers in Aging Neuroscience. 2019;11:34.
20. Lauretti E, Dincer O, Praticò D. Regional and temporal miRNAs expression profile in a transgenic mouse model of tauopathy: implication for its pathogenesis. Mol Psychiatry. 2020;10
21. RAY, C. George; RYAN, Kenneth James. Sherris medical microbiology. McGraw-Hill, 2010:260-261
22. Gasca, A. T., Vicente, M. I. A., & Díaz, A. T. Las misteriosas y peligrosas enfermedades priónicas o encefalopatías espongiformes transmisibles: nuevos y revolucionarios conceptos en patología. Manual formativo de ACTA. 2006;(41):23-33.
23. Guo, T., Noble, W., & Hanger, D. P.Roles of tau protein in health and disease. Acta neuropathologica, 2017;133(5):665-704.
24. Davidowitz EJ, Krishnamurthy PK, Lopez P, Jimenez H, Adrien L, Davies P, et al. In Vivo Validation of a Small Molecule Inhibitor of Tau Self-Association in htau Mice. JAD. 2020;73(1):147-61.
25. Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett, 1993; 336(3):417–424.
26. Cavallini, A., Brewerton, S., Bell, A., Sargent, S., Glover, S., Hardy, C., Moore, R., Calley, J., Ramachandran, D., Poidinger, M., Karran, E., Davies, P., Hutton, M., Szekeres, P., & Bose, S. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem, 2013;288(32), 23331–23347
27. Saito T, Oba T, Shimizu S, Asada A, Iijima KM, Ando K. Cdk5 increases MARK4 activity and augments pathological tau accumulation and toxicity through tau phosphorylation at Ser262. Hum Mol Genet. 2019;28(18):3062-71.
28. Ma, X., Liu, L., & Meng, J. MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer’s disease. Neuroscience Letters. 2017;661:57–62
29. Wang, Y., Veremeyko, T., Wong, A. H.-K., El Fatimy, R., Wei, Z., Cai, W., & Krichevsky, A. M. (2017). Downregulation of miR-132/212 impairs S-nitrosylation balance and induces tau phosphorylation in Alzheimer’s disease. Neurobiology of Aging, 51, 156–166. doi:10.1016/j.neurobiolaging.2016.12.015
30. Estrada Rodríguez, A. E., & Zomosa Signoret, V. C. Papel de la agregación del péptido Beta amiloide en la enfermedad de Alzheimer. Revista de Educación Bioquímica. 2017;36(1):2-11.
31. Giordano, R. Análisis bioquímico en el diagnóstico temprano de Alzheimer. Proteínas Tau y Beta-amiloide.Anuario fundación Dr. J.R Villavicencio. 2006; No XIV: 40-41
32. Glenner G, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984;120:885–890.
33. Kim DE, Priefer R. Therapeutic Potential of Direct Clearance of the Amyloid-β in Alzheimer's Disease. Brain Sci. 2020;10(2):E93.
34. Hillen H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front Neurosci. 2019 Nov 7;13 [Citado el 16 de abril de 2020] Disponible en: https://www.frontiersin.org/articles/10.3389/fnins.2019.01154/ful
35. Manzano-León, N., & Mas-Oliva, J. Estrés oxidativo, péptido β-amiloide y enfermedad de Alzheimer. Gaceta médica de México. 2006;142(3):229-238.
36. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends in Pharmacological Sciences. 1991;12:383-8.
37. Gulisano W, Maugeri D, Baltrons MA, Fà M, Amato A, Palmeri A, et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. JAD. 2018;64(s1):S611-S631.
38. Kolata G. Down syndrome--Alzheimer's linked. Science. 1985;230(4730):1152-3.
39. Majláth Z, Toldi J, Vécsei L. The potential role of kynurenines in Alzheimer’s disease: pathomechanism and therapeutic possibilities by influencing the glutamate receptors. J Neural Transm. 2014;121(8):881-9.
40. Powers RK, Culp-Hill R, Ludwig MP, Smith KP, Waugh KA, Minter R, et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun. 2019;10:4766.
41. Roy ER, Wang B, Wan Y, Chiu G, Cole A, Yin Z, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. Journal of Clinical Investigation. 2020 ;130(4):1912-30.
42. Shinohara, M., Tachibana, M., Kanekiyo, T., & Bu, G. . Role of LRP1 in the pathogenesis of Alzheimer’s disease: evidence from clinical and preclinical studies. Journal of lipid research. 2017;58(7):1267-1281.
43. Van Gool, B., Storck, S. E., Reekmans, S. M., Lechat, B., Gordts, P. L., Pradier, L., ... & Roebroek, A. J.. LRP1 Has a Predominant Role in Production over Clearance of Aβ in a Mouse Model of Alzheimer’s Disease. Molecular neurobiology. 2019;6(10):7234-7245.
44. Bilousova, T., Melnik, M., Miyoshi, E., Gonzalez, B. L., Poon, W. W., Vinters, H. V., ... & Albay III, R.. Apolipoprotein E/Amyloid-β Complex Accumulates in Alzheimer Disease Cortical Synapses via Apolipoprotein E Receptors and Is Enhanced by APOE4. The American journal of pathology. 2019;189(8):1621-1636.
45. Grimmer, T., Goldhardt, O., Yakushev, I., Ortner, M., Sorg, C., Diehl-Schmid, J., ... & Miners, S. Associations of Neprilysin Activity in CSF with Biomarkers for Alzheimer’s Disease. Neurodegenerative Diseases. 2019;19(1):43-50.
46. Campos, C. R., Kemble, A. M., Niewoehner, J., Freskgård, P. O., & Urich, E. Brain Shuttle Neprilysin reduces central Amyloid-β levels. Plos one. 2020;15(3):e0229850.
47. Ries, M., Loiola, R., Shah, U. N., Gentleman, S. M., Solito, E., & Sastre, M. The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-β peptide. Journal of neuroinflammation. 2016;13(1):234.
48. Kurochkin, I. V., Guarnera, E., & Berezovsky, I. N. Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends in pharmacological sciences, 2018;39(1):49-58.
49. Quan, Q., Qian, Y., Li, X., & Li, M. Pioglitazone Reduces β Amyloid Levels via Inhibition of PPARγ Phosphorylation in a Neuronal Model of Alzheimer’s Disease. Frontiers in aging neuroscience,2019;11:178.
50. Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, et al. Enhanced clearance of A in brain by sustaining the plasmin proteolysis cascade. Proceedings of the National Academy of Sciences. 2008 Jun 24;105(25):8754-9.[Citado el 18 y 19 de Abril de 2020] Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2438386/
51. Ries M, Sastre M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front Aging Neurosci. 2016 Jul 5;8: [Citado el 18 y 19 de Abril de 2020] Disponible en: https://www.frontiersin.org/articles/10.3389/fnagi.2016.00160/full
52. Cai M, Jung I, Kwon H, Cho E, Jeon J, Yun J, et al. Spinosin Attenuates Alzheimer’s Disease-Associated Synaptic Dysfunction via Regulation of Plasmin Activity. Biomolecules & Therapeutics. 2020 Mar 1;28(2):131-6 [Citado el 18 y 19 de Abril de 2020] Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7059816/
53. Hemming, M. L., & Selkoe, D. J. Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. Journal of Biological Chemistry. 2005;280(45): 37644-37650.
54. Hu, J., Igarashi, A., Kamata, M., & Nakagawa, H. Angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide (Aβ); retards Aβ aggregation, deposition, fibril formation; and inhibits cytotoxicity. Journal of Biological Chemistry. 2001;276(51):47863-47868.
55. Itakura E, Chiba M, Murata T, Matsuura A. Heparan sulfate is a clearance receptor for aberrant extracellular proteins. Journal of Cell Biology. 2020;219(3):e201911126.
56. Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, et al. Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiol Aging. 2011;32(5):763-77.
57. Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh MF. Impact of HMGB1, RAGE, and TLR4 in Alzheimer's Disease (AD): From Risk Factors to Therapeutic Targeting. Cells. 2020;9(2):E383.
58. Prasad K. AGE-RAGE stress: a changing landscape in pathology and treatment of Alzheimer's disease. Mol Cell Biochem. 2019;459(1-2):95-112.
59. Weller J, Budson A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 2018;7:1161.
60. Hatami, A., Albay, R., Monjazeb, S., Milton, S., & Glabe, C. Monoclonal antibodies against Aβ42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. Journal of Biological Chemistry. 2014;289(46):32131-32143.
61. Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alz Res Therapy. 2016;8(1):18.
62. Fuller JP, Stavenhagen JB, Christensen S, Kartberg F, Glennie MJ, Teeling JL. Comparing the efficacy and neuroinflammatory potential of three anti-abeta antibodies. Acta Neuropathol. 2015;130(5):699-711.
63. Crenezumab | ALZFORUM [Internet]. Alzforum.org. 2020 [citado el 16 de Abril 2020]. Disponible de: https://www.alzforum.org/therapeutics/crenezumab
64. Schwarz AJ, Sundell KL, Charil A, Case MG, Jaeger RK, Scott D, et al. Magnetic resonance imaging measures of brain atrophy from the EXPEDITION 3 trial in mild Alzheimer's disease. Alzheimer's & Dementia: Translational Research & Clinical Interventions. 2019;5(1):328-37.
65. Schneider L. A resurrection of aducanumab for Alzheimer's disease. The Lancet Neurology. 2020;19(2):111-2.
66. Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369(4):341-50
67. Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, et al. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015;72(11):1324-33
68. Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, et al. Randomized Trial of Verubecestat for Prodromal Alzheimer's Disease. N Engl J Med. 2019;380(15):1408-20.
69. Wessels AM, Tariot PN, Zimmer JA, Selzler KJ, Bragg SM, Andersen SW, et al. Efficacy and Safety of Lanabecestat for Treatment of Early and Mild Alzheimer Disease. JAMA Neurol. 2020;77(2):199.
70. Scratch Lanabecestat: This BACE Inhibitor Doesn’t Work in Symptomatic AD, Either | ALZFORUM [Internet]. Alzforum.org. 2020 [cited 16 April 2020]. Available from: https://www.alzforum.org/news/research-news/scratch-lanabecestat-bace-inhibitor-doesnt-work-symptomatic-ad-either
71. Lue L, Yan S, Stern D, Walker D. Preventing Activation of Receptor for Advanced Glycation Endproducts in Alzheimer's Disease. CDTCNSND. 2005;4(3):249-66.
72. Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer's Disease. J Prev Alzheimers Dis. 2018;5(2):149-54.
73. Zhao Y, Alexandrov P, Jaber V, Lukiw W. Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer’s Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7). Genes. 2016;7(12):116.
74. Madadi S, Schwarzenbach H, Saidijam M, Mahjub R, Soleimani M. Potential microRNA-related targets in clearance pathways of amyloid-β: novel therapeutic approach for the treatment of Alzheimer’s disease. Cell Biosci. 2019;9:91.
75. Amakiri N, Kubosumi A, Tran J, Reddy PH. Amyloid Beta and MicroRNAs in Alzheimer’s Disease. Front Neurosci. 2019;13:430.
76. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9-19
77. Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biology. 2018;14:450-64.
78. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239-47.
79. Singh A, Kukreti R, Saso L, Kukreti S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019;24(8):E 1583.
80. Shukla D, Mandal PK, Tripathi M, Vishwakarma G, Mishra R, Sandal K. Quantitation of in vivo brain glutathione conformers in cingulate cortex among age‐matched control, MCI, and AD patients using MEGA‐PRESS. Hum Brain Mapp. 2020;41(1):194-217.
81. Pocernich CB, Butterfield DA. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim Biophys Acta. 2012;1822(5):625-30.
82. González-Acosta CA, Escobar MI, Casanova MF, Pimienta HJ, Buriticá E. Von Economo Neurons in the Human Medial Frontopolar Cortex. Front Neuroanat. 2018;12:64.
83. Cauda F, Torta DME, Sacco K, D’Agata F, Geda E, Duca S, et al. Functional anatomy of cortical areas characterized by Von Economo neurons. Brain Struct Funct. 2013;218(1):1-20.
84. Santillo AF, Nilsson C, Englund E. von Economo neurons are selectively targeted in frontotemporal dementia. Neuropathol Appl Neurobiol. 2013;39(5):572-9.
85. Rogalski EJ, Gefen T, Shi J, Samimi M, Bigio E, Weintraub S, et al. Youthful memory capacity in old brains: anatomic and genetic clues from the Northwestern SuperAging Project. J Cogn Neurosci. 2013;25(1):29-36.
86. Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):211-20.
87. Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256(8):1205-14.
88. Huang W, Zhou Y, Tu L, Ba Z, Huang J, Huang N, et al. TDP-43: From Alzheimer’s Disease to Limbic-Predominant Age-Related TDP-43 Encephalopathy. Front Mol Neurosci. 2020;13:26.
89. Davis SA, Itaman S, Khalid-Janney CM, Sherard JA, Dowell JA, Cairns NJ, et al. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neuroscience Letters. 2018;678:8-15
90. Davis SA, Gan KA, Dowell JA, Cairns NJ, Gitcho MA. TDP-43 expression influences amyloidβ plaque deposition and tau aggregation. Neurobiology of Disease. 2017;103:154-62
91. Gu J, Hu W, Tan X, Qu S, Chu D, Gong CX, et al. Elevation of casein kinase 1ε associated with TDP-43 and tau pathologies in Alzheimer's disease. Brain Pathol. 2020;30(2):283-97.
92. Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117(2):125-36.
93. Gu J, Wu F, Xu W, Shi J, Hu W, Jin N, et al. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Research. 2017;45(10):6177-93.
94. Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127(6):811-24.
95. Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22(8):869-78.
96. Wang P, Deng J, Dong J, Liu J, Bigio EH, Mesulam M, et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019;15(5):e1007947.
97. APOE apolipoprotein E [Homo sapiens (human)] - Gene - NCBI [Internet]. Ncbi.nlm.nih.gov. 2020 [citado el 27 de Abril de 2020]. Disponible en: https://www.ncbi.nlm.nih.gov/gene/348
98. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63(3):287-303.
99. Uddin, M. S., Kabir, M. T., Al Mamun, A., Abdel-Daim, M. M., Barreto, G. E., & Ashraf, G. M. (2018). APOE and Alzheimer’s Disease: Evidence Mounts that Targeting APOE4 may Combat Alzheimer’s Pathogenesis. Molecular Neurobiology. doi:10.1007/s12035-018-1237-z
100. Suidan, G. L., & Ramaswamy, G. Targeting Apolipoprotein E for Alzheimer’s Disease: An Industry Perspective. International Journal of Molecular Sciences. 2019;20(9):2161.
101. Konttinen H, Cabral-da-Silva MeC, Ohtonen S, Wojciechowski S, Shakirzyanova A, Caligola S, et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Reports. 2019;13(4):669-83.
102. Lin Y, Seo J, Gao F, Feldman HM, Wen H, Penney J, et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018;98(6):1141-1154.e7.
103. Wolfe CM, Fitz NF, Nam KN, Lefterov I, Koldamova R. The Role of APOE and TREM2 in Alzheimer's Disease-Current Understanding and Perspectives. Int J Mol Sci. 2018;20(1):E81.
104. McQuade A, Blurton-Jones M. Microglia in Alzheimer's Disease: Exploring How Genetics and Phenotype Influence Risk. J Mol Biol. 2019;431(9):1805-17.
105. Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213(5):667-75.
106. Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol Neurodegeneration. 2018;13(1):29
107. Atagi Y, Liu C, Painter MM, Chen X, Verbeeck C, Zheng H, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem. 2015;290(43):26043-50
108. Yamazaki Y, Zhao N, Caulfield TR, Liu C, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501-18.
109. Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020;11(1):667.
110. Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25(11):1680-3.
111. Williams T, Borchelt DR, Chakrabarty P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer's disease. Mol Neurodegener. 2020;15(1):8.
112. Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron. 2017;96(5):1013-1023.e4.
113. Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO, Michaelson DM. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J Alzheimers Dis. 2016;54(3):1219-33.
114. Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287(8):5253-66.
115. [Internet]. Genenames.org. 2020 [citado el 7 de Mayo de 2020]. Disponible de: https://www.genenames.org/data/gene-symbol-report/#!/hgnc_id/HGNC:11138
116. Bonini NM, Giasson BI. Snaring the function of alpha-synuclein. Cell. 2005;123(3):359-61.
117. Emamzadeh FN. Alpha-synuclein structure, functions, and interactions. J Res Med Sci. 2016;21:29.
118. Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90(23):11282-11286.
119. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388(6645):839-40.
120. Korff A, Liu C, Ginghina C, Shi M, Zhang J. α-Synuclein in cerebrospinal fluid of Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis. 2013;36(4):679-88.
121. Twohig D, Nielsen HM. α-synuclein in the pathophysiology of Alzheimer's disease. Mol Neurodegener. 2019;14(1):23.
122. Vasili E, Dominguez-Meijide A, Outeiro TF. Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front Mol Neurosci. 2019;12:107.
123. Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener. 2020;15(1):19.
124. Aprile FA, Källstig E, Limorenko G, Vendruscolo M, Ron D, Hansen C. The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci Rep. 2017;7(1):9039.
125. Bhasne K, Sebastian S, Jain N, Mukhopadhyay S. Synergistic Amyloid Switch Triggered by Early Heterotypic Oligomerization of Intrinsically Disordered α-Synuclein and Tau. Journal of Molecular Biology. 2018;430(16):2508-20.
126. Lu J, Zhang S, Ma X, Jia C, Liu Z, Huang C, et al. Structural basis of the interplay between α-synuclein and Tau in regulating pathological amyloid aggregation. J Biol Chem. 2020;295(21):7470-7480.
127. Snyder, H. M., Corriveau, R. A., Craft, S., Faber, J. E., Greenberg, S. M., Knopman, D., Carrillo, M. C. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s & Dementia. 2015;11(6):710–717.
128. Korczyn AD, Vakhapova V, Grinberg LT. Vascular dementia. J Neurol Sci. 2012;322(1-2):2-10.
129. Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet. 2015;6:112.
130. Jefferson AL, Cambronero FE, Liu D, Moore EE, Neal JE, Terry JG, et al. Higher Aortic Stiffness Is Related to Lower Cerebral Blood Flow and Preserved Cerebrovascular Reactivity in Older Adults. Circulation. 2018;138(18):1951-62.
131. Qiu C, Winblad B, Viitanen M, Fratiglioni L. Pulse pressure and risk of Alzheimer disease in persons aged 75 years and older: a community-based, longitudinal study. Stroke. 2003;34(3):594-9.
132. McGrath ER, Beiser AS, DeCarli C, Plourde KL, Vasan RS, Greenberg SM, et al. Blood pressure from mid‐ to late life and risk of incident dementia. Neurology. 2017;89(24):2447-54.
133. Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science. 2019;365(6450):eaav9518.
134. Breteler, M. M. Vascular risk factors for Alzheimer’s disease: Neurobiology of Aging. 2000;21(2): 153–160.
135. Klohs J. An Integrated View on Vascular Dysfunction in Alzheimer’s Disease. Neurodegener Dis. 2019;19(3-4)109-127.
136. Buchman AS, Tanne D, Boyle PA, Shah RC, Leurgans SE, Bennett DA. Kidney function is associated with the rate of cognitive decline in the elderly. Neurology. 200922;73(12):920-7.
137. Kuo, Y., Li, C., Sung, J. et al. Risk of dementia in patients with end-stage renal disease under maintenance dialysis—a nationwide population-based study with consideration of competing risk of mortality. Alz Res Therapy. 2019;11(1):31.
138. Liu Y, Xiang Y, Wang Y, Jiao S, Wang Q, Bu X, et al. Association Between Serum Amyloid-Beta and Renal Functions: Implications for Roles of Kidney in Amyloid-Beta Clearance. Mol Neurobiol. 2015;52(1):115-9.
139. Takae, K., Hata, J., Ohara, T., Yoshida, D., Shibata, M., Mukai, N., et al. Albuminuria Increases the Risks for Both Alzheimer Disease and Vascular Dementia in Community‐Dwelling Japanese Elderly: The Hisayama Study. J Am Heart Assoc. 2018;7(2):e006693
140. Deckers K, Camerino I, van Boxtel MP, Verhey FR, Irving K, Brayne C, et al. Dementia risk in renal dysfunction: A systematic review and meta-analysis of prospective studies. Neurology. 2017;88(2):198-208.
141. Kurella Tamura M, Pajewski NM, Bryan RN, Weiner DE, Diamond M, Van Buren P, et al. Chronic kidney disease, cerebral blood flow, and white matter volume in hypertensive adults. Neurology. 2016;86(13):1208-16.
142. Simões E Silva AC, Miranda AS, Rocha NP, Teixeira AL. Neuropsychiatric Disorders in Chronic Kidney Disease. Front Pharmacol. 2019;10:932.
143. Laske C, Sopova K, Stellos K. Platelet Activation in Alzheimer’s Disease: From Pathophysiology to Clinical Value. CVP. 2012;10(5):626-30.
144. Stellos K, Panagiota V, Kögel A, Leyhe T, Gawaz M, Laske C. Predictive Value of Platelet Activation for the Rate of Cognitive Decline in Alzheimer's Disease Patients. J Cereb Blood Flow Metab. 2010;30(11):1817-20.
145. Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14(1):28-39.
146. Zamolodchikov D, Chen Z, Conti BA, Renné T, Strickland S. Activation of the factor XII-driven contact system in Alzheimer’s disease patient and mouse model plasma. Proc Natl Acad Sci USA. 2015;112(13):4068-73.
147. Chen Z, Revenko AS, Singh P, MacLeod AR, Norris EH, Strickland S. Depletion of coagulation factor XII ameliorates brain pathology and cognitive impairment in Alzheimer disease mice. Blood. 2017;129(18):2547-56.
148. Cortes-Canteli M, Kruyer A, Fernandez-Nueda I, Marcos-Diaz A, Ceron C, Richards AT, et al. Long-Term Dabigatran Treatment Delays Alzheimer’s Disease Pathogenesis in the TgCRND8 Mouse Model. Journal of the American College of Cardiology. 2019;74(15):1910-23.
149. Vemuri P, Knopman DS, Lesnick TG, Przybelski SA, Mielke MM, Graff-Radford J, et al. Evaluation of Amyloid Protective Factors and Alzheimer Disease Neurodegeneration Protective Factors in Elderly Individuals. JAMA Neurol. 2017;74(6):718.

