

Sección C: Ingenierías
Comportamiento del SDS localizado en la región interfacial del sistema agua/n-octano. Un estudio usando dinámica molecular
ACI Avances en Ciencias e Ingenierías
Universidad San Francisco de Quito, Ecuador
ISSN: 1390-5384
ISSN-e: 2528-7788
Periodicidad: Bianual
vol. 8, núm. 1, 2015
Recepción: 02 Marzo 2016
Publicación: 13 Diciembre 2016
Autor de correspondencia: jgparra2@uc.edu.ve

Resumen: En este trabajo, se establecio una metodolog´ ´ıa usando dinamica molecular tipo NVT y NPT para´ estimar las propiedades interfaciales y el comportamiento del dodecil sulfato de sodio en la region´ interfacial de los sistemas vac´ıo/agua y agua/n-octano. La tension interfacial fue estimada usando el´ modelo propuesto por Kirkwood y Buff [23]. Para ello, los modelos de energ´ıa potencial GROMOS53A6 y SPC fueron usados para describir las moleculas de n-octano, SDS y agua, respectivamente.´ A su vez, los espesores de pel´ıcula interfacial fueron determinados usando los criterios 10-90 y 90-90 sobre los perfiles de densidad de los sistemas simulados. Ademas, el´ area por mol´ ecula fue´ calculado usando la variacion de la presi´ on superficial en funci´ on de la concentraci´ on del surfactante.´ Para el sistema agua/n-octano, la tension interfacial obtenida fue de 52.20 mN/m. En los sistemas´ vac´ıo/SDS/agua, el area por mol´ ecula del SDS fue obtenida con dos procedimientos diferentes. Los´ valores estimados fueron 53.3 A˚ 2 y 54.3 A˚ 2, respectivamente. Por ultimo, para los sistemas agua/n-´ octano y agua/sds/n-octano los espesores de pel´ıcula interfacial aumentan en funcion del n´ umero de´ moleculas de surfactantes presentes en la regi´ on interfacial, con un m´ ´ınimo de tension interfacial´ de 4.94 mN/m y un valor maximo de espesor de pel´ ´ıcula de 19.22 A. El˚ area por mol´ ecula para el´ sistema agua/SDS/n-octano fue de 46.5 A˚ 2 para un m´ınimo de tension interfacial de 4.94 mN/m. Los´ resultados obtenidos en este trabajo son consistentes con datos medidos por experimentacion.´
Palabras clave: Dinamica molecular, espesor de película interfacial, tension interfacial.
Abstract: In this paper, a methodology using molecular dynamics NVT and NPT was established to estimate the interfacial properties and behavior of sodium dodecyl sulfate in the interfacial region of the vacuum/water and water/n-octane systems. The interfacial tension was estimated using the model proposed by Kirkwood and Buff [23]. For that, the GROMOS-53A6 and SPC models were used to describe the n-octane, SDS and water molecules, respectively. In turn, to determine the interfacial film thickness the criteria 10-90 and 90-90 were applied to the density profiles of simulated systems. Also, the area per molecule was calculated using the variation of the surface pressure in function of concentration of the surfactant. For water/n-octane system, the interfacial tension obtained was of 52.20 mN/m. Moreover, the values of area per molecule of SDS in water estimated with two procedure differents. The obtained values were 53.3 A˚ 2 and 54.3 A˚ 2, respectively . Finally, for water/noctane and water/sds/n-octane systems the thickness of the interfacial film increase in function of the number of molecules of surfactants present in the interfacial region with a minimum interfacial tension of 4.94 mN/m and a maximum value of film thickness of 19.22 A. Area per molecule of the˚ water/SDS/n-octane system was 46.5 A˚ 2 for this interfacial tension. The results are consistent with experimental values.
Keywords: Molecular Dynamic, interfacial film thickness, interfacial tension.
Introducción
Los surfactantes, son moleculas anfifılicas que tienen una cabeza polar de naturaleza hidrofılica y una cadena hidrocarbonada tipo lipofılica [1, 2]. Estas moleculas, tienen la capacidad de reducir la tension interfacial de las interfaces vapor/lıquido, lıquido/lıquido y lıquido/solido.
La adsorcion de surfactantes en la region interfacial depende de sus propiedades anfifılicas unicas, las cuales son conocidas como el balance entre las fuerzas hidrofılicas y lipofılicas de la cabeza polar y la cadena lipofılica. El comportamiento de los surfactantes ubicados en las regiones interfaciales juegan un papel importante en muchas aplicaciones como detergencia, flotacion mineral, dispersion de solidos, recuperacion de crudos, dispersion de nanopartıculas, entre otros. Todas estas aplicaciones han motivado diversos estudios para describir agregados y monocapas de surfactantes en diferentes interfaces [3]. A nivel experimental, el dodecil sulfato de sodio (SDS, C12.25OSO
) es uno de los surfactantes ionicos mas utilizados en los estudios de agregacion micelar, estabilidad de espumas y formacion de monocapas. A su vez, el SDS tiene alto valor de solubilidad en agua lo cual expresa su alto nivel hidrofılico [4]. Experimentalmente, se ha estudiado la adsorcion de SDS en las regiones interfaciales aire/aceite, aire/agua [5–7] y agua/grafito usando espectroscopıa de frecuencia vibracional, reflactancia de neutrones y microscopıa de fuerza atomica con la finalidad de estimar la orientacion molecular de los surfactantes´ en la region interfacial [8, 9].´
Con el desarrollo computacional, las tecnicas de simulacion como dinamica molecular ha sido muy utilizadas para estudiar el auto ensamblaje de surfactantes en las regiones interfaciales aire/agua y aire/aceite a nivel atom´ıstico [10–14]. En estos trabajos, se determinaron las tensiones interfaciales, las presiones superficiales y algunas propiedades estructurales. Sin embargo, existen pocos trabajos donde se haya determinado la energıa necesaria para la formacion de la interfaz del sistema vacıo/SDS/agua [15] . Adicionalmente, las propiedades estructurales y el mecanismo de la agregacion del SDS en agua han sido estudiadas con dinamica molecular [16, 17].
Por tal motivo, en este trabajo, se determinaron las propiedades interfaciales (presion superficial, pelıcula interfacial y area por molecula) de los sistemas vacıo/SDS/agua y agua/SDS/n-octano, con la finalidad de profundizar en el comportamiento del surfactante SDS desde el punto de vista molecular.
Metodología empleada
Modelos de energía potencial
En simulaciones con dinamica molecular, los sistemas moleculares son descritos por un modelo de energıa potencial denominado force field [18]. El modelo de energıa potencial incluye los terminos enlazantes y no enlazantes, los cuales incluyen las fuerzas electrostaticas y de van der Waals [18]. Por lo tanto, la energıa de interaccion total entre moleculas puede ser escrita como,
Donde, Eintra, Einter . Etotal son las energ´ıas intramolecular, intermolecular y total, respectivamente.
En este trabajo, las moleculas de SDS y n-octano fueron descritas usando el modelo de energıa potencial GROMOS53A6 [18]. Los parametros de este modelo de energıa potencial fueron generados con el software Automated force field Topology Builder (ATB) [19]. En el force field GROMOS53A6, los terminos asociados a las interacciones no enlazantes fueron calculados mediante la ecuacion 2,
 (2)
(2)Donde, Aij, Bij, q., q. and rij son los parametros de energıa, cargas atomicas y distancia entre partıculas . y ., respectivamente.
Para el agua, el modelo de punto de carga simple (Simple Point Charge, SPC) fue usado en nuestra simulacion (SPC model) [20]. En este modelo, la distancia OH es 0.100 nm y el´ angulo HOH es 109.47´ .. Ademas, existe una´ unica interacci´ on de Lennard-Jones entre los sitios del ox´ ´ıgeno. Los parametros´ de Lennard-Jones y cargas atomicas en el modelo SPC son´ .(O) = 0,317 nm,
kJ/mol, q(H) = 0,4100e y q(O) = −0,8200e. El modelo SPC permite predecir resultados exactos de las propiedades del agua como la densidad y la difusion en fase lıquida.
Perfiles de densidad y pelıcula interfacial
El perfil de densidad de un sistema interfacial describe la no homogeneidad de una interfaz y permite localizar la region interfacial y el espesor de la misma. Para una interfaz lıquido/lıquido, la region interfacial comienza, donde la densidad de ambos lıquidos disminuye con respecto al seno del lıquido puro. Generalmente, los perfiles de densidad de una partıcula tipo i, a lo largo de la direccion perpendicular a´ la interfaz se describe por la ecuacion 3:´
 (3)
(3)Donde, ρ.(.) es el perfil de densidad a lo largo del eje z, hN.(.)i es el numero de part´ ´ıculas tipo i entre . − z/2 y . + z/2 en el tiempo t [21]. En este trabajo, ∆. fue escogido a un valor igual a 0.01 nm.
Tension interfacial
La propiedad macroscopica m´ as importante para definir un sistema interfacial es la tensi´ on superficial.´ Los metodos usados para calcular la tensi´ on superficial son basados sobre la definici´ on mec´ anica [22–´ 26]. La primera forma expl´ıcita desarrollada por Kirkwood and Buff expresa a los tensores de presion´ como una funcion derivada de la energ´ ´ıa potencial [23]. En este caso, la tension superficial es definida´ por las ecuacion 4:´
 (4)
(4)Donde, P. . P. son los componentes normal y tangencial de la presion, respectivamente.´ L. es la dimension de la celda de simulaci´ on a lo largo del eje z. El componente normal´ P. es igual a Pzz, mientras el componente tangencial P. es dado por 1.2(Pxx . Pyy). Los tensores de presion´ Pxx, Pyy . Pzz, desde el punto de vista molecular, son definidos de forma general mediante la ecuacion 5:´
 (5)
(5)Donde . es el tensor unitario, k. es la constante de Boltzmann, . es la temperatura, y . = N/Ves la densidad en numero. Los t´ erminos´ . y . representan las direcciones X, Y, y Z. En esta ecuacion 5,´ rij es el vector entre el centro de masa de la molecula i y j. El t´ ermino´ Fij es la fuerza intermolecular entre moleculas i y j, el cual se expresa como la suma de todas las fuerzas interactuantes entre estas moleculas´ [27].
Generalmente, en dinamica molecular la tensi´ on interfacial se calcula usando el tensor presi´ on promedio.´ En este caso, la tension interfacial es determinada a lo largo del eje z mediante la ecuaci´ on 6:´
 (6)
(6)
Sistemas simulados
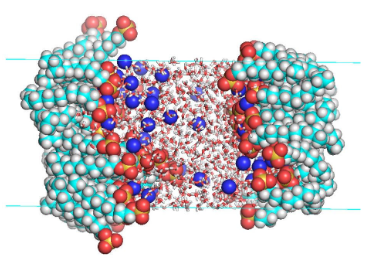
En este estudio se utilizaron dos tipos de sistemas. Para los sistemas vac´ıo/SDS/agua se construyeron celdas periodicas rectangulares de longitudes Lx = Ly = 3 nm y Lz = 30 nm, con una capa de agua´ constituida por 1000 moleculas ubicada en el centro de la celda a lo largo del eje z. Las monocapas de´ surfactantes conten´ıan 9, 12, 16, 20, 25, 30 moleculas de SDS y se colocaron a los extremos de la capa´ de agua como se puede observar en la figura 1.
Las dimensiones de las celdas periodicas de agua y n-octano fueron de 5x5x4´ nm.. Estas celdas periodi-´ cas se unieron formando una caja rectangular de 5x5x8 nm.. El numero de mol´ eculas de SDS presentes´ en las monocapas fueron 9, 12, 16, 20, 25 y 36. Adicionalmente, las moleculas de SDS y n-octano han´ sido descritas con el modelo de energ´ıa potencial GROMOS-53A6 [28, 29]. Para el caso del SDS, los parametros usados corresponden al modelo de Berkowitz y colaboradors [30, 31]. En cambio, el modelo´ SPC fue usado para simular las moleculas de agua. Al final de las simulaciones, los sistemas estaban´ confinados en cajas rectangulares de 4x4x8.2 nm..
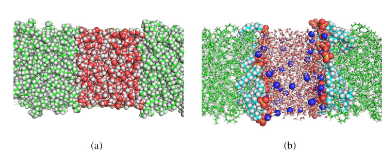
Seguidamente, los sistemas agua/n-octano y agua/SDS/n-octano fueron construidos usando 188 molecu-´ las de n-octano y 1640 moleculas de agua. En la figura 2, se muestra un ejemplo de los modelos cons-´ tru´ıdos. La construccion de los sistemas simulados se realiz´ o usando las herramientas genbox, genconf´ y editconf del programa Groningen Machine for Chemical Simulations (GROMACS) version 4.5.5 [32–35]
Condiciones de la simulaciones.
Todas las simulaciones se realizaron con el programa Gromacs 4.5.5. Los sistemas fueron periodicos´ en las coordenadas XYZ. La temperatura y presion usada fue de 300 K y 1 atm, respectivamente. El´ metodo de Berendsen fue utilizado para controlar la temperatura. La constante de acoplamiento para el´ termostato de Berendsen fue de 0.1 ps [36].
Las velocidades iniciales de las part´ıculas fueron generadas usando una distribucion Maxweliana a 300´ K y las ecuaciones de movimiento se integraron usando el algoritmo leapfrog con un paso del tiempo de 1 fs. Las interacciones de Lennard-Jones se calcularon usando un radio de interaccion de 1.40 nm y las´ interacciones electrostaticas fueron calculadas usando el procedimiento de mallado de Ewald (PME) con´ un radio de interaccion de 1.30 nm [37].´
Los sistemas vac´ıo/SDS/agua/SDS/vac´ıo fueron relajados usando el metodo de minimizaci´ on gradiente´ conjugado. Luego, la configuracion final obtenida se le aplicaron dos simulaciones de din´ amica mole-´ cular tipo NVT. Las simulaciones tuvieron un lapso de 10 ns a 300 K. Con las trayectorias obtenidas de la segunda simulacion tipo NVT, se estim´ o la tensi´ on interfacial, la energ´ ´ıa de formacion de la inter-´ faz (EFI) y el espesor de la pel´ıcula interfacial de cada uno de los sistemas. Para calcular el espesor de pel´ıcula interfacial se utilizo el criterio 10-90 por ser el m´ as simple. Las trayectorias fueron almacenadas´ cada 1000 fs para determinar los promedios de las propiedades.
Los sistemas agua/n-octano y agua/sds/n-octano fueron relajados usando el metodo de minimizaci´ on´ gradiente conjugado y steep descent. Las monocapas de surfactante fueron colocadas sobre las superficies del agua y estos sistemas SDS/agua/SDS fueron relajados usando una dinamica molecular tipo NVT de´ 500 ps. A las configuraciones finales obtenidas, se le colocaron las capas de n-octano en ambos lados de la celda. Estos sistemas fueron relajados haciendo una minimizacion con el m´ etodo steep descent.´ Luego, la configuracion final obtenida fue la usada para comenzar la din´ amica molecular. Despu´ es del´ proceso de minimizacion, se realizaron simulaciones de din´ amica molecular tipo NPT de 10 ns a 300´ K. Posteriormente, se realizo una segunda simulaci´ on tipo NVT de 10 ns a las mismas condiciones de´ temperatura. Finalmente, se almacenaron las trayectorias cada 1 ps. Los ultimos 5 ns de la din´ amica´ molecular tipo NVT se utilizaron para determinar las propiedades.
La tension interfacial fue estimada con el modelo de Kirwood-Buff usando los tensores de presi´ on local´ y el espesor de pel´ıcula interfacial se determino usando el criterio 10-90 y 90-90.´
Resultados y Discusión
Perfiles de densidad de los sistemas vacıo/SDS/agua.
Inicialmente, con las trayectorias obtenidas de las dinamicas molecular tipo NVT, se determinaron los´ perfiles de densidad de los sistemas vac´ıo/SDS/agua. En la figura 3, se muestran los perfiles de densidad de los sistemas agua/SDS/vac´ıo simulados.
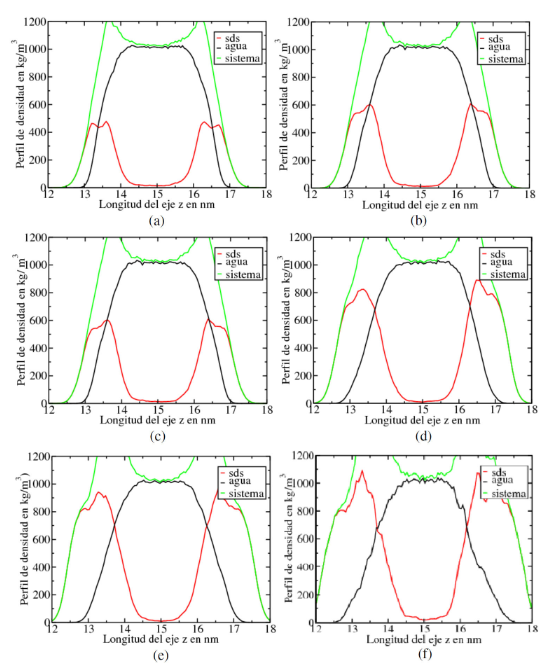
Como era de esperarse, se obtuvo un incremento en el espesor del perfil de densidad del SDS en el vac´ıo a medida que aumenta el numero de mol´ eculas de SDS en la monocapa. Este comportamiento se debe a´ la mayor presencia de cadenas lipof´ılicas en los sistemas estudiados.
A su vez, se observa un solapamiento entre los perfiles de densidad del agua y el SDS, lo cual es debido a las interacciones tipo ion-dipolo de los iones sodio y el grupo hidrof´ ´ılico sulfato presente en el surfactante con las moleculas de agua ubicadas en la regi´ on interfacial.´
Adicionalmente, el perfil de densidad del agua se muestra invariable en el seno del l´ıquido. Tambien´ se observa que el perfil de densidad del agua se distorsiona a medida que aumenta la concentracion de´ SDS. Este fenomeno ocurre en la regi´ on interfacial debido al aumento de las interacciones moleculares´ entre el SDS y el agua. En este trabajo, el sistema vac´ıo/SDS/agua constituido por monocapas con 20 moleculas de SDS representa un superficie de agua saturada con dicho surfactante. Luego, aplicando el´ criterio 10-90 sobre los perfiles de densidad del agua en los sistemas vac´ıo/SDS/agua, se determinaron los espesores de pel´ıcula interfacial. Estos valores se muestran en la tabla 1.
| Moleculas de SDS´ | Area por´ | Pel´ıcula |
| en la monocapa | molecula en´ A˚ 2 | interfacial en A˚ |
| 9 | 100.00 | 7.42 |
| 12 | 75.00 | 7.88 |
| 16 | 56.25 | 8.45 |
| 20 | 45 | 9.45 |
| 25 | 36 | 10.46 |
| 30 | 30 | 11.12 |
Para el sistema saturado vac´ıo/SDS/agua, el espesor de pel´ıcula interfacial fue de 9.45 A (tabla 1) .˚ Esta magnitud de espesor de pel´ıcula interfacial se debe al aumento en las interacciones de la parte hidrof´ılica del SDS con el agua en la region interfacial. Si suponemos que el espesor de pel´ ´ıcula del agua pura es de 4.5 A , entonces el espacio ocupado por una monocapa de SDS en un sistema saturado es de˚ 4.95 A para el sistema vac˚ ´ıo/SDS/agua. De igual manera, el espesor de pel´ıcula interfacial aumenta en funcion de la cantidad de mol´ eculas de SDS en la regi´ on interfacial.´
Tension interfacial y energıa de formacion de la interfaz de los sistemas vacıo/SDS/agua.
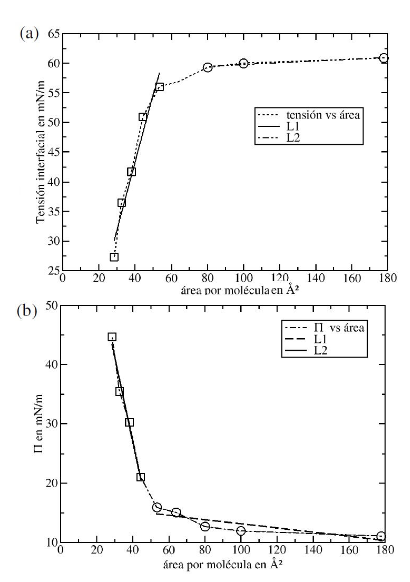
Para este trabajo se estimo la tensi´ on interfacial de los sistemas vac´ ´ıo/SDS/agua en funcion del´ area´ ocupada por molecula de surfactante. Esto se puede apreciar en la figura 4.
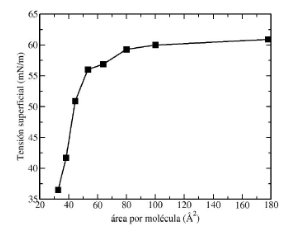
Aqu´ı, se encontro que cuando el´ area por mol´ ecula es mayor a 50´ A˚ 2, la tension interfacial de los sistemas´ vac´ıo/SDS/agua se hace constante. En cambio, cuando el area interfacial es menor a 50´ A˚ 2 por molecula´ se encuentra un cambio de pendiente en la curva de tension superficial en funci´ on del´ area por mol´ ecula.´ Estos resultados muestran que la superficie del agua esta saturada con SDS cuando el valor de area´ superficial es de 50 A˚ 2 . En cambio, cuando el area por mol´ ecula es muy grande, la tensi´ on interfacial´ tiende al valor del agua pura. Para calcular el area por mol´ ecula se utiliz´ o el m´ etodo de regresi´ on lineal´ sobre las curvas de tension superficial y presi´ on superficial de los sistemas vac´ ´ıo/SDS/agua. Para calcular la presion superficial,´ Π, se utilizo la ecuaci´ on 7:´
 (7)
(7)Donde γvacio/agua, es la tension del agua pura y´ γsistema es la tension del sistema vac´ ´ıo/SDS/agua. En las figuras 5(a) y 5(b), se muestran las curvas de tension interfacial y presi´ on superficial en funci´ on´ del area por mol´ ecula para el sistema vac´ ´ıo/SDS/agua.
En este estudio usando dinamica molecular y el cambio de pendiente de la curva de presi´ on superficial,´ se obtuvo un valor teorico de´ area por mol´ ecula de 53.3´ A˚ 2. En cambio, usando la curva de tension super-´ ficial en funcion del´ area por mol´ ecula se obtuvo un valor de 54.3´ A˚ 2. Usando medidas experimentales de tension interfacial en agua fue reportado un valor de´ area por mol´ ecula para el SDS de 53´ A˚ 2 [2]. Esto indica que la metodolog´ıa aplicada es consistente para predecir las propiedades interfaciales del surfactante SDS en agua. A su vez, es importante destacar que la seleccion de un modelo de energ´ ´ıa potencial con buenos parametros moleculares permite predecir adecuadamente las propiedades interfaciales de los´ sistemas simulados.
Adicionalmente, se determino la energ´ ´ıa para la formacion de la interfaz (EFI) de los sistemas vac´ ´ıo/SDS/agua. Para ello, se construyeron dos sistemas moleculares. El primer sistema fue constru´ıdo con una molecula´ de SDS aislada en una celda periodica de dimensiones 4x4x30´ nm3 . Luego, el segundo sistema consist´ıa de una capa con 1000 moleculas de agua ubicada en el fondo de una celda peri´ odica con las mismas di-´ mensiones. A los dos sistemas se les realizaron dinamicas moleculares tipo NVT de 2ns a 300 K para´ calcular la energ´ıa total del surfactante SDS aislado, (Esurf ), y la energ´ıa total de la capa de agua, (Eagua ). A su vez, la energ´ıa total del sistema vac´ıo/SDS/agua, (Esistema), se calculo para las configuraciones´ obtenidas en cada tiempo t de la simulacion. La ecuaci´ on 8, muestra como se calcula la energ´ ´ıa necesaria para la formacion de la interfaz por mol´ ecula de surfactante (EFI):´
 (8)
(8)Donde, nsurf, corresponde al numero de mol´ eculas de surfactantes presentes en la monocapa. La EFI se´ grafico en funci´ on del´ area por mol´ ecula de SDS. La figura 6, muestra la variaci´ on de dicha energ´ ´ıa en funcion del´ area por mol´ ecula de SDS.´
Cuando la superficie de agua se satura con moleculas de SDS, el sistema vac´ ´ıo/SDS/agua se hace mas´ estable (figura 6). Por lo general, la energ´ıa de formacion de la interfaz vac´ ´ıo/SDS/agua es una medida de las interacciones moleculares por surfactante que surge de la insercion de dichas mol´ eculas en la regi´ on´ interfacial vac´ıo/agua.
Para un area por mol´ ecula de 80´ A˚ 2, el sistema muestra un cambio de pendiente. Aqu´ı, la energ´ıa necesaria para la formacion de la interfaz fue de -189.79 kJ/mol. Esto sugiere un aumento en las interacciones´ moleculares entre las moleculas de SDS que se acomodan sobre la superficie de agua. Dicha estabiliza-´ cion puede ser debida a las interacciones dispersivas entre las cadenas lipof´ ´ılicas de los surfactantes que se acomodan sobre la superficie del l´ıquido.
Perfiles de densidad de los sistemas agua/SDS/n-octano y agua/n-octano
En esta parte, se determinaron los perfiles de densidad de los sistemas agua/SDS/n-octano y agua/noctano usando dinamica molecular tipo NPT a 300 K. Para la estimaci´ on de los espesores de pel´ ´ıcula interfacial, se construyeron los perfiles de densidad usando 600 planos divisorios de la celda periodica a´ lo largo del eje z y la separacion entre cada plano fue de 0,01´ A. En la figura 8, se muestra el perfil de˚ densidad del sistema n-octano/agua obtenido despues de la segunda simulaci´ on NPT de 5 ns a 300 K y 1´ atm.
Los espesores de pel´ıcula interfacial se determinaron usando los criterios 10-90 y 90-90. El criterio 10-90 se refiere al aumento del perfil de densidad desde un 10 por ciento hasta un 90 por ciento y el criterio 90-90, corresponde al espesor medido entre los l´ımites 90 por ciento y 90 por ciento de los perfiles de densidad de ambas capas inmiscibles. Usando el criterio 10-90, fueron obtenidos dos espesores de pel´ıcula interfacial usando las capas de hidrocarburo y agua.
Para el sistema agua/n-octano, los perfiles de densidad a lo largo del eje z son consistentes con las densidades experimentales de los l´ıquidos puros. La densidad promedio obtenida para este sistema fue (820.84 ± 0.40) Kg.m..
Generalmente, la densidad de los sistemas bifasicos oscila entre las densidades del agua y del hidrocar-´ buro. En este trabajo, el modelo SPC usado para describir las moleculas de agua estima adecuadamente´ la densidad del sistema agua/n-octano estudiado.
Para el perfil del n-octano, se encuentran ciertas fluctuaciones de la densidad cerca de la region interfacial´ lo cual es debido al rearreglo de las moleculas de n-octano por la repulsi´ on que presenta con las mol´ eculas´ de agua. En cambio, el perfil de densidad del agua se encuentra sin perturbaciones debido a que el modelo usado en la simulacion es muy r´ ´ıgido.
Usando el criterio 10-90, se encuentran dos valores de espesores de pel´ıcula interfacial usando los perfiles de densidad del agua y del n-octano. Con el perfil de densidad del agua, el espesor de pel´ıcula obtenido fue de 0.395 nm.
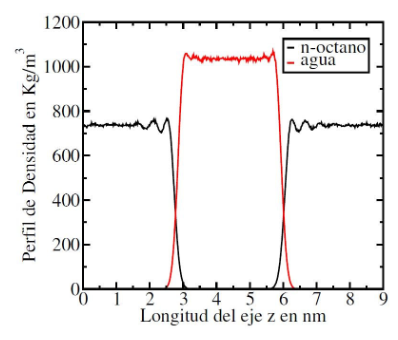
Figura 8: Perfil de densidad del sistema agua/n-octano obtenido con GROMACS 4.5.5.
A su vez, utilizando el perfil de densidad del n-octano, el espesor de pel´ıcula interfacial fue de 0.415 nm. Estos resultados son consistentes con valores reportados ya mencionados anteriormente. Por ejemplo, Mitrinovic et al. reportaron un valor experimental de 0.350 nm para el sistema n-hexano/agua [41]. Tambien, Riedleder et al. reportaron una amplitud interfacial de 0.380 nm para el sistema n-heptano/agua´ usando el criterio 10-90 [42]. En base a esto, encontramos que usando este criterio, los espesores de pel´ıcula interfacial son de mayor magnitud usando los perfiles de los hidrocarburos.
En cambio, usando el criterio 90-90, se encontro un´ unico valor de espesor de pel´ ´ıcula interfacial para los sistemas estudiados. En este caso para el sistema agua/n-octano, la magnitud fue de 0.495 nm. Generalmente, usando este criterio, los espesores de pel´ıcula interfacial son sobreestimados.
Luego, se colocaron monocapas de surfactantes SDS en la region interfacial del sistema agua/n-octano.´ En este caso, se estudio el efecto de la concentracion de surfactante sobre esta propiedad interfacial´ denominada espesor de pel´ıcula. En la tabla 3, se muestran los valores obtenidos de espesor de pel´ıcula interfacial usando los criterios 10-90 y 90-90.
Se muestra que a medida que se adiciona moleculas de surfactantes a la regi´ on interfacial se genera un´ aumento de espesor de pel´ıcula interfacial. Usando el criterio 10-90 sobre la capa de agua, se encuentra magnitudes mas coherentes de espesor de pel´ ´ıcula interfacial. En cambio, usando la capa de n-octano hay un aumento considerable de los espesores de pel´ıcula. Lo mismo ocurre usando el criterio 90-90. Sin embargo, en todos los casos la pel´ıcula interfacial aumenta en funcion de la concentraci´ on de surfactante´ en la region interfacial (ver tabla 3).´
Tabla 3: Espesores de pel´ıcula interfacial obtenidos para los sistemas agua/sds/n-octano usando los criterios 10-90 y 90-90
| Moleculas de SDS´ | Criterio 10-90 en A˚ | Criterio 10-90 en A˚ | Criterio 90-90 en A˚ |
| agua | n-octano | ||
| 9 | 4.37 | 9.30 | 11.04 |
| 12 | 5.01 | 10.46 | 13.01 |
| 16 | 5.43 | 11.71 | 14.77 |
| 20 | 5.77 | 11.90 | 15.49 |
| 25 | 6.24 | 12.38 | 16.69 |
| 36 | 8.55 | 12.34 | 19.22 |
Desde el punto de vista teorico, el aumento de espesor de pel´ ´ıcula implica una mayor estabilidad del sistema agua/SDS/n-octano. Generalmente, el SDS adicionado forma una monocapa autoensamblada en la region interfacial n-octano/agua y la parte lipof´ ´ılica de este penetra la capa de hidrocarburo. A su vez, la parte hidrof´ılica interacciona fuertemente con el agua debido a la afinidad que presentan entre s´ı. Esto ocasiona la permeacion del n-octano y agua en la membrana de surfactante autoensamblada y un aumento´ de los espesores de pel´ıcula medidos en las capas de n-octano y agua. De igual manera, con el criterio 90-90, se obtienen altos valores de espesor de pel´ıcula interfacial. De hecho, el aumento en el espesor de pel´ıcula en la capa de agua es debido al grupo hidrof´ılico. En cambio para la capa de hidrocarburo es debido a la cadena lipof´ılica.
En la tabla 4, se muestra el aumento en la pel´ıcula debido al grupo hidrof´ılico y lipof´ılico presentes en el surfactante dodecil sulfato de sodio. La contribucion al espesor de pel´ ´ıcula interfacial total debido al grupo hidrof´ılico es menor en comparacion a la contribuci´ on del grupo lipof´ ´ılico. A su vez, a medida que la concentracion de surfactante aumenta la contribuci´ on de cada parte del surfactante aumenta. Para el´ sistema completamente saturado con surfactante, el cual corresponde a 36 moleculas en la monocapa, el´ espesor de pel´ıcula obtenido fue de 14.27 A.˚
Tabla 4: Aumento de la pel´ıcula interfacial (.) en nm obtenidos usando los criterios 10-90 y 90-90. El agua fue descrita usando el modelo SPC-E.
| Moleculas de SDS´ | Grupo hidrof´ılico | Grupo lipof´ılico | Total |
| A˚ | A˚ | A˚ | |
| 9 | 0.42 | 5.15 | 6.09 |
| 12 | 1.06 | 6.31 | 8.06 |
| 16 | 1.48 | 7.56 | 9.82 |
| 20 | 1.82 | 7.75 | 10.54 |
| 25 | 2.29 | 8.23 | 11.74 |
| 36 | 4.6 | 8.19 | 14.27 |
Conclusiones
En este trabajo, se lograron determinar las propiedades interfaciales de los sistemas vac´ıo/SDS/agua y agua/SDS/n-octano usando dinamica molecular tipo NVT y NPT y los modelos de energ´ ´ıa potencial GROMOS-53A6 y SPC para describir las moleculas de n-octano, SDS y agua, respectivamente.´
El valor de area por mol´ ecula del SDS en agua estimado fue de 53.3´ A˚ . . A su vez, segun la curva de´ energ´ıa necesaria para la formacion de la interfaz se presenta un cambio de pendiente para un´ area de´ 80 A˚ . y un valor de energ´ıa de -189.79 kJ/mol. Estos resultados muestran que las interacciones del SDS con el agua son efectivas cuando la superficie esta saturada con dicho surfactante. A su vez, el espesor de´ la pel´ıcula interfacial aumenta en funcion de la concentraci´ on de surfactante SDS ubicado en la regi´ on´ interfacial.
Ademas, se encontr´ o que el n´ umero de mol´ eculas de agua presentes en la primera capa de hidrataci´ on´ aumenta en funcion del n´ umero de mol´ eculas de SDS ubicadas en la regi´ on interfacial. En el punto de´ saturacion de la superficie del agua, el n´ umero de mol´ eculas de agua en la primera capa de hidrataci´ on´ corresponde a 19.74 moleculas.´
Tambien, se determin´ o la capacidad del SDS para reducir la tensi´ on interfacial en un sistema agua/n-´ octano. Los perfiles de densidad del sistema agua/n-octano a lo largo del eje z son consistentes con las densidades experimentales de los l´ıquidos puros. La densidad promedio obtenida para este sistema fue (820.84 ± 0.40) Kg.m.. Adicionalmente, para los sistemas agua/n-octano y agua/sds/n-octano se pudo precisar que los espesores de pel´ıcula interfacial aumentan en funcion del n´ umero de mol´ eculas de´ surfactantes presentes en la region interfacial con un m´ ´ınimo de tension interfacial de 4.94 mN/m y un´ valor maximo de espesor de pel´ ´ıcula de 19.22 A usando los force field GROMOS-53A6 y SPC.˚ Particularmente, estos resultados son debidos a la buena interaccion que existe entre el grupo hidrof´ ´ılico del surfactante y las moleculas de agua, lo cual genera una disminuci´ on de la tensi´ on interfacial y un´ aumento del espesor de pel´ıcula.
Los resultados obtenidos de las simulaciones fueron consistentes con valores obtenidos experimentalmente.
Referencias
Myers, D. (2002). Surface activity and surfactant structures. Surfaces, Interfaces, and Colloids: Principles and Applications, Second Edition, 21-39.
[2] Rosen, M. J., & Kunjappu, J. T. (2012). Surfactants and interfacial phenomena. John Wiley & Sons.
[3] Myers, D. (2005). Surfactant science and technology. John Wiley & Sons.
[4] Holmberg, K. (Ed.). (2002). Handbook of applied surface and colloid chemistry (Vol. 1). New York: Wiley.
[5] Rehfeld, S. J. (1967). Adsorption of sodium dodecyl sulfate at various hydrocarbon-water interfaces. The Journal of Physical Chemistry, 71(3), 738-745, DOI: 10.1021/j100862a039.
[6] Saien, J. & Akbari, S. (2006). Interfacial Tension of Toluene + Water + Sodium Dodecyl Sulfate from (20 to 50) ◦C and pH between 4 and 9. J. Chem. Eng. Data, 51(5), 1832-1835, DOI: 10.1021/je060204g.
[7] Hansen, M. & Short, D. (1990). Optimization study of octane-in-water emulsions by sedimentation field-flow fractionation. Journal of Chromatography, 517, 333-344.
[8] Tummala, N. R., & Striolo, A. (2008). Role of counterion condensation in the self-assembly of SDS surfactants at the water-graphite interface. The Journal of Physical Chemistry B, 112(7), 1987-2000.
[9] Watry, M. R., & Richmond, G. L. (2000). Comparison of the adsorption of linear alkanesulfonate and linear alkylbenzenesulfonate surfactants at liquid interfaces. Journal of the American Chemical Society, 122(5), 875-883.
[10] Zhao, T., Xu, G., Yuan, S., Chen, Y., & Yan, H. (2010). Molecular dynamics study of alkyl benzene sulfonate at air/water interface: effect of inorganic salts. The Journal of Physical Chemistry B, 114(15), 5025-5033.
[11] Shi, L., Tummala, N. R., & Striolo, A. (2010). C12E6 and SDS surfactants simulated at the vacuumwater interface. Langmuir, 26(8), 5462-5474.
[12] Yan, H., Guo, X. L., Yuan, S. L., & Liu, C. B. (2011). Molecular dynamics study of the effect of calcium ions on the monolayer of SDC and SDSn surfactants at the vapor/liquid interface. Langmuir, 27(10), 5762-5771.
[13] Shi, L., Tummala, N. R., & Striolo, A. (2010). C12E6 and SDS surfactants simulated at the vacuumwater interface. Langmuir, 26(8), 5462-5474.
[14] Bresme, F., Chacon, E., Mart´ ´ınez, H., & Tarazona, P. (2011). Adhesive transitions in Newton black films: A computer simulation study. The Journal of chemical physics, 134(21), 214701.
[15] Jang, S. S., & Goddard, W. A. (2006). Structures and properties of newton black films characterized using molecular dynamics simulations. The Journal of Physical Chemistry B, 110(15), 7992-8001.
[16] Bruce, C. D., Senapati, S., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulations of sodium dodecyl sulfate micelle in water: the behavior of water. The Journal of Physical Chemistry B, 106(42), 10902-10907.
[17] Bruce, C. D., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulation of sodium dodecyl sulfate micelle in water: micellar structural characteristics and counterion distribution. The Journal of Physical Chemistry B, 106(15), 3788-3793.
[18] Oostenbrink, C., Villa, A., Mark, A. E., & Van Gunsteren, W. F. (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force field parameter sets 53A5 and 53A6. Journal of computational chemistry, 25(13), 1656-1676.
[19] Malde, A. K., Zuo, L., Breeze, M., Stroet, M., Poger, D., Nair, P. C., ... & Mark, A. E. (2011). An automated force field topology builder (ATB) and repository: version 1.0. Journal of chemical theory and computation, 7(12), 4026-4037.
[20] Hermans, J., Berendsen, H. J., Van Gunsteren, W. F., & Postma, J. P. (1984). A consistent empirical potential for water–protein interactions. Biopolymers, 23(8), 1513-1518.
[21] Geysermans, P., Elyeznasni, N., & Russier, V. (2005). Layered interfaces between immiscible liquids studied by density-functional theory and molecular-dynamics simulations. The Journal of chemical physics, 123(20), 204711.
[22] Rowlinson, J. S., & Widom, B. (2013). Molecular theory of capillarity. Courier Corporation.
[23] Kirkwood, J. G., & Buff, F. P. (1949). The statistical mechanical theory of surface tension. The Journal of Chemical Physics, 17(3), 338-343.
[24] Irving, J. H.; Kirkwood, J. G. The Statistical Mechanical Theory of Transport Processes. IV. The Equations of Hydrodynamics. J. Chem.Phys. 1950, 18, 817-829.
[25] Irving, J. H., & Kirkwood, J. G. (1950). The statistical mechanical theory of transport processes. IV. The equations of hydrodynamics. The Journal of chemical physics, 18(6), 817-829.
[26] Walton, J. P. R. B., & Gubbins, K. E. (1985). The pressure tensor in an inhomogeneous fluid of non-spherical molecules. Molecular Physics, 55(3), 679-688.
[27] Neyt, J. C., Wender, A., Lachet, V., Ghoufi, A., & Malfreyt, P. (2014). Quantitative Predictions of the Interfacial Tensions of Liquid–Liquid Interfaces through Atomistic and Coarse Grained Models. Journal of chemical theory and computation, 10(5), 1887-1899.
[28] Van Gunsteren, W. F., & Berendsen, H. J. C. (1987). Groningen molecular simulation (GROMOS) library manual. Biomos, Groningen, 24(682704), 13.
[29] Oostenbrink, C., Villa, A., Mark, A. E., & Van Gunsteren, W. F. (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force field parameter sets 53A5 and 53A6. Journal of computational chemistry, 25(13), 1656-1676.
[30] Bruce, C. D., Senapati, S., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulations of sodium dodecyl sulfate micelle in water: the behavior of water. The Journal of Physical Chemistry B, 106(42), 10902-10907.
[31] Bruce, C. D., Berkowitz, M. L., Perera, L., & Forbes, M. D. (2002). Molecular dynamics simulation of sodium dodecyl sulfate micelle in water: micellar structural characteristics and counterion distribution. The Journal of Physical Chemistry B, 106(15), 3788-3793.
[32] Hess, B., Kutzner, C., Van Der Spoel, D., & Lindahl, E. (2008). GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of chemical theory and computation, 4(3), 435-447.
[33] Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. (2005). GROMACS: fast, flexible, and free. Journal of computational chemistry, 26(16), 1701-1718.
[34] Lindahl, E., Hess, B., & Van Der Spoel, D. (2001). GROMACS 3.0: a package for molecular simulation and trajectory analysis. Molecular modeling annual, 7(8), 306-317.
[35] Berendsen, H. J., van der Spoel, D., & van Drunen, R. (1995). GROMACS: a message-passing parallel molecular dynamics implementation. Computer Physics Communications, 91(1), 43-56.
[36] Berendsen, H. J., Postma, J. V., van Gunsteren, W. F., DiNola, A. R. H. J., & Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. The Journal of chemical physics, 81(8), 3684-3690.
[37] Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., & Pedersen, L. G. (1995). A smooth particle mesh Ewald method. The Journal of chemical physics, 103(19), 8577-8593.
[38] Zhao, T., Xu, G., Yuan, S., Chen, Y., & Yan, H. (2010). Molecular dynamics study of alkyl benzene sulfonate at air/water interface: effect of inorganic salts. The Journal of Physical Chemistry B, 114(15), 5025-5033.
[39] Xu, J., Zhang, Y., Chen, H., Wang, P., Xie, Z., Yao, Y., ... & Zhang, J. (2013). Effect of surfactant headgroups on the oil/water interface: An interfacial tension measurement and simulation study. Journal of Molecular Structure, 1052, 50-56.
[40] Chen, Y., & Xu, G. (2013). Improvement of Ca 2+-tolerance by the introduction of EO groups for the anionic surfactants: Molecular dynamics simulation. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 424, 26-32.
[41] Mitrinovic, D. M., Tikhonov, A. M., Li, M., Huang, Z., & Schlossman, M. L. (2000). Noncapillarywave structure at the water-alkane interface. Physical review letters, 85(3), 582.
[42] Riedleder, A. J., Kentish, S. E., Perera, J. M., & Stevens, G. W. (2007). Structural Investigation of a Water/n-Heptane Interface: A Molecular Dynamics Study. Solvent Extraction and Ion Exchange, 25(1), 41-52.
[43] Zhang, Y., Feller, S. E., Brooks, B. R., Pastor, R. W. (1995). Computer simulation of liquid/liquid interfaces. I. Theory and application to octane/water. J. Chem. Phys., 23, 10252-10266.
Notas de autor
jgparra2@uc.edu.ve

