

SARS-CoV-2: structure, replication and pathophysiological mechanisms related to COVID-19
Gaceta Médica Boliviana, vol. 43, no. 2, 2020
Universidad Mayor de San Simón
Articulos de Revision
Received: 07 July 2020
Accepted: 16 September 2020

Abstract: SARS-CoV2 is the cause of severe acute respiratory syndrome, a disease that has also been named COVID-19, it was reported in late 2019 as a new betacoronavirus in people who were exposed in a seafood market in Wuhan, China. The virus since that date has been spreading rapidly causing a pandemic, and affecting various countries to a greater or lesser extent. Currently, there is a wide range of information about the virus and the disease; knowledge about the pathophysiology and the way in which this entity should be managed has been changing over time. Despite the interest that has been generated in the pathophysiological mechanisms of the disease and its complications, these have not been fully understood. This article provides a systematized review of the structure, replication and pathophysiological aspects related to SARS-CoV2, which has caused a high rate of morbidity and mortality in the population worldwide.
Keywords: SARS-CoV-2, COVID-19, betacoronavirus, physiopathological mechanisms, morbidity.
Resumen: SARS-CoV2 es causante del síndrome respiratorio agudo severo, enfermedad que ha sido también nombrada COVID-19, fue notificado a fines del año 2019 como un nuevo betacoronavirus en personas expuestas en un mercado de mariscos en Wuhan, China. El virus desde esa fecha se ha ido propagando rápidamente provocando una pandemia, y afectando a diversos países en mayor o menor magnitud. Actualmente existe información variada difundida sobre el virus y la enfermedad; los conocimientos sobre la fisiopatología y la manera en la que debe ser gestionada esta entidad han ido cambiando a través de tiempo. A pesar del interés que se ha generado en los mecanismos fisiopatológicos de la enfermedad y sus complicaciones, estos no se han llegado a descifrar totalmente. Mediante el presente artículo se hace una revisión sistematizada de la estructura, replicación y aspectos fisiopatológicos relacionados con SARS-CoV2, que ha provocado un elevado índice de morbimortalidad en la población a nivel mundial.
Palabras clave: SARS-CoV-2, COVID-19, betacoronavirus, mecanismos fisiopatológicos, morbimortalidad.
The new severe acute respiratory syndrome coronavirus (SARS-CoV2) is a virus belonging to the family Coronaviridae, subfamily Coronavirinae1; It causes severe acute respiratory syndrome or also called COVID-19 which was reported in late 2019 as a new betacoronavirus in people exposed at a seafood market in Wuhan, China1,2. Outbreaks of severe acute respiratory syndrome (SARS) in 2002/2003 and Middle East respiratory syndrome (MERS) in 2012, also caused by coronavirus species have demonstrated the possibility of animal-to-human and human-to-human transmission2,3. SARS-CoV-2 shares about 96.2% sequence homology with bat coronavirus and this virus is currently believed to have been introduced into humans by an as yet unidentified animal intermediate and then spread from human to human3.
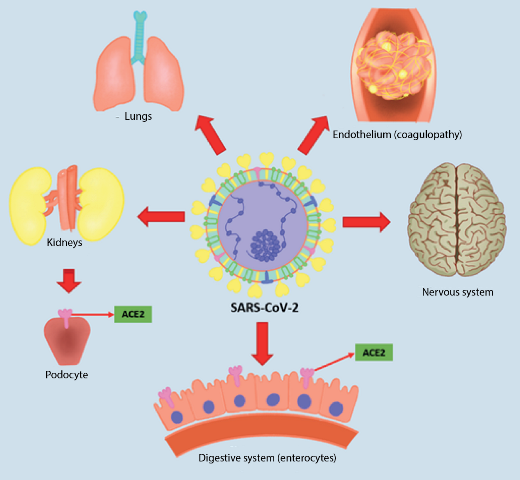
Symptoms of COVID-19, a disease so named by Dr. Tedros Adhanom Ghebreyesus, Director General of the World Health Organization (WHO) on February 11, 2020, varies depending on whether it is a mild, moderate or severe form of the disease4. In mild cases of COVID-19, symptoms may include thermal rises, dry cough, malaise, myalgia, anosmia and ageusia; some patients have gastrointestinal symptoms, such as anorexia, nausea, vomiting and diarrhea1. Severe cases of COVID-19 occur mostly in patients with chronic underlying disease such as cardiovascular disease, diabetes mellitus, chronic kidney disease and obesity among others; however, it has also been reported in patients without comorbidity of all ages5,6. These patients may have serious complications such as acute respiratory distress syndrome (ARDS) with dyspnoea and hypoxaemia, lymphopenia, central or peripheral nervous system disorders, renal failure, heart failure, hepatic failure, haemopathies and shock5-8. This article will attempt to elucidate the molecular basis of the pathophysiology of COVID-19, which will help us to understand not only the disease, but also its complications and the mechanisms necessary to reach the multi-organ failure that occurs in several reported cases of this disease (See figure 1).
Literature review
For this article, an exhaustive systematic search of primary sources of information was carried out in online search engines and scientific indexes, such as: PubMed, Elsevier, Scielo, MedLine, etc.
The primary sources themselves correspond to published and pre-published scientific articles (Review Article, Clinical Guidelines, Case Report, Case Series and Clinical Trials) which will be mentioned in the final bibliography.
Topic development and discussion
General structure of SARS-CoV-2
Four genera of coronaviruses are recognised: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus2;. Genealogical examination of SARS-CoV-2 revealed that it belongs to the coronavirus genus betacoronavirus4.
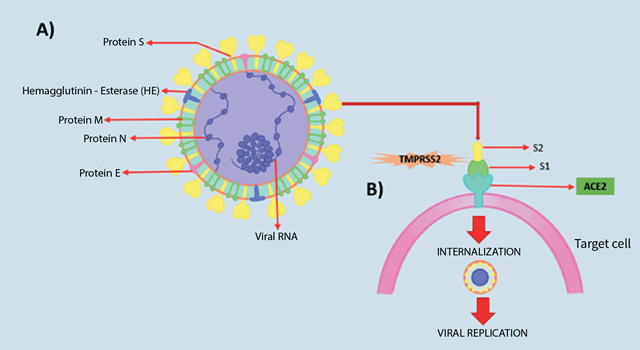
Coronaviruses get their name from the electron microscopic appearance of their virions, which resemble a solar corona (with surface projections) due to their surface proteins9. Structurally, coronaviruses are spherical viruses measuring 80-160 nanometres in diameter, with a lipid bilayer envelope and containing a single-stranded, positively polarised RNA (ssRNA) genome between 27 and 30 kilobases in length9. The SARS-CoV-2 virus genome encodes 5 structural proteins, which are encoded within the 3’ end of the viral genome9, 10, 11:
Glycoprotein S (spike): Trimeric glycoprotein S is a class I fusion protein and mediates binding to the host receptor. Glycoprotein S is cleaved by a host cell furin-like protease into two separate polypeptides called S1 and S2. S1 constitutes the large receptor-binding domain of the S protein, while S2 forms the stalk of the spike molecule
Protein E (envelope): The transmembrane protein E has an N-terminal ectodomain and a C-terminal endodomain and has ion channel activity. The ion channel activity in the SARS-CoV E protein is not required for viral replication, but may be required for pathogenesis. It facilitates virus assembly and release.
Protein M (membrane): The most abundant structural protein in the virion. It is a small protein with three transmembrane domains. It was suggested that the M protein exists as a dimer in the virion, and can adopt two different conformations, allowing it to promote membrane curvature and bind to the nucleocapsid. This protein is thought to give the virion its shape.
Protein N (nucleocapsid): Composed of two separate domains, an N-terminal domain and a C-terminal domain, both capable of binding to RNA in vitro, but each one utilises different mechanisms to bind RNA. The N protein is also highly phosphorylated, and it has been suggested that phosphorylation triggers a structural change that enhances affinity for viral versus non-viral RNA. The N protein binds to the viral genome in a bead-on-a-string conformation.
Haemagglutinin esterase (HE): Present in a subset of betacoronaviruses. The protein acts as a haemagglutinin, binds sialic acids on surface glycoproteins and contains acetyl esterase activity. These activities are thought to enhance protein S-mediated cell entry and virus spread across the mucosa.
Among these five proteins, the most important are protein N and protein S, where the former helps the virus to develop the capsid and the complete viral structure properly and the latter helps the virus to bind to host cells11 (See figure 2).
General pathomechanism of SARS-CoV-2
SARS-CoV-19 (causative agent of severe acute respiratory syndrome in 2002/2003) and SARS-CoV-2 (causative agent of COVID-19) have been reported to have a similar type of receptor. Protein S binds directly to the angiotensin-converting enzyme 2 (ACE2) receptor on host target cells 10,11. The ACE2 receptor is expressed in several organs of the human body, mainly in the lungs, kidneys and intestine which represent the main targets of the coronavirus11, 12. The binding affinity of SARS-CoV-2 to the ACE2 receptor is 10 to 20 times higher compared to SARS-CoV-112.
Most pathogenic viruses have been found to contain a furin-like cleavage site in the S protein, which is not present in SARS-CoV-1 but is present in SARS-CoV-2; this process requires cellular serine proteases (TMPRSS2), which allow cleavage of the S protein, regulating the whole mechanism and thus enhancing viral fusion with host cell membranes13. Protein S thus possesses two functional subunits S1 (N-terminal) and S2 (C-terminal) mentioned above; the former mediates the binding of the virus to the host cell membrane by recognising a receptor on the cognate cell, while the latter promotes fusion of the 2 cell membranes, and is involved in viral entry14.
SARS-CoV-2 Replication
Once the virus has entered the host cell, it initiates the replication process; the virus genome contains a large replicase gene that will give rise to non-structural proteins (Nsps), followed by structural and accessory genes9,15. The replicase gene encodes two open reading frames (ORFs), rep1a and rep1b, which are translated into two polypeptides (pp1a and pp1ab); these polypeptides are processed by two viral proteases: 3C-type protease (3CLpro) and papain-type protease. Cleavage produces 15 or 16 viral Nsps that assemble into a large membrane-bound complex and exhibit multiple enzymatic activities15.
The positive-strand RNA genome is used as a template to produce the negative strand. Enzymes encoded by the replicase gene use the negative RNA as a template to develop overlapping messenger RNA (mRNA) segments that are translated into structural proteins.11 It is thought that the manufacture of these individual RNA molecules could promote recombination events between viral genomes and genetic diversity.9
During the replication process within the human host, the N protein of the virus binds to the genome, while the M protein associates with the membranes of the endoplasmic reticulum (ER). Subsequently, messenger RNA and nucleocapsid proteins combine to form virions11. The viral particles are directed to the endoplasmic reticulum-Golgi apparatus intermediate complex and from this compartment the vesicles containing the virions are directed to fuse with the plasma membrane, thus assembling the complete viral particles which, when released, go on to infect new cells16.
Inflammatory response caused by SARS-CoV-2
A “cytokine storm “17 has been suggested in infection with SARS-CoV-1 and various respiratory viruses. In the case of SARS-CoV-2, similar inflammatory mechanisms leading to the clinical deterioration of patients have also been suggested18,19. This response is defined by low levels of type I and III interferons juxtaposed with elevated chemokines and high expression of interleukin-6 (IL-6)19.
During viral replication, the host cell mediated by a family of intracellular pattern recognition receptors (PRRs) detects aberrant RNA structures that often form during viral replication; these receptors oligomerise and trigger the activation of downstream transcription factors: interferon regulatory factors (IRFs) and nuclear factor κB (NF-κB)20. The activation of these regulatory factors particularly activates two antiviral programs.
The first is mediated by transcription of type I and III interferons (IFN-I and IFN-III, respectively) and subsequent up-regulation of IFN-stimulated genes. The second involves the recruitment and coordination of specific subsets of leukocytes, which is characterised by the secretion of chemokines19,21. It has been postulated that the host response to SARS-CoV-2 is unable to launch a robust IFN-I and III response while inducing high levels of chemokines necessary to recruit effector cells19,22.
Proinflammatory cytokines and chemokines that were elevated during COVID-19 infection include tumour necrosis factor α (TNF- α), interleukin 1β (IL-1β), IL-6, granulocyte colony-stimulating factor (G-CSF), interferon gamma-induced protein 10 (IP-10), monocyte chemoattractant protein 1 (MCP-1) and macrophage inflammatory proteins 1- α (MIP 1- α)23.
SARS-CoV-2 and SARS-CoV-2 and severe acute respiratory distress syndrome.
SARS-CoV-2 enters human type 2 pneumocytes via the ACE-2 receptor, then initiates the replication process described above13. Xu et al. performed histopathological studies with biopsy samples of lung tissue in patients with COVID-19 and have demonstrated the presence of diffuse alveolar damage with the formation of hyaline membranes, presence of mononuclear cells and macrophages infiltrating air spaces with diffuse thickening of the alveolar wall24. Viral particles have also been observed in type II pneumocytes by electron microscopy23. These pathological alterations can lead to severe acute respiratory distress syndrome (SDRA), characterised by severe hypoxaemia, acute onset of bilateral infiltrates that have been described in a “ground-glass” pattern on X-rays, and pulmonary oedema that cannot be explained by fluid overload or heart failure5.
In the pathogenesis of SDRA, there is an epithelial lesion with increased permeability in the alveolocapillary membrane, as well as diffuse damage to the alveolar cells that leads to fluid accumulation, inactivation of surfactant produced by type 2 pneumocytes and subsequent formation of the hyaline membrane, which is considered impermeable to gas exchange; These changes occurring at the pulmonary level would cause an increase in the work of breathing in the patient with signs of respiratory distress, and there would also be an intrapulmonary shunt with increased blood flow that would also affect gas exchange with the appearance of refractory hypoxaemia in these patients25.
SARS-CoV-2 and coagulopathy
Coagulopathy is a common feature of SARS-CoV-2 infection, often characterised by increased D-dimer and fibrin as a common laboratory finding26.
D-dimer is the end product of fibrin degradation that serves as a serological indicator of the activation of coagulation and the fibrinolytic system27 .
In patients with severe COVID-19 it has been described that there is direct local vascular and endothelial injury leading to microvascular clot formation and angiopathy; there is also a hypercoagulable state with hyperfibrinogenemia and the possibility of large vessel thrombosis or major thromboembolic sequelae, including pulmonary thromboembolism (TEP), which is reported in up to 20-30% of intensive care unit (UCI) patients28.
The coagulation cascade is activated in viral infections such as COVID-19; there is an imbalance between procoagulant and anticoagulant homeostatic mechanisms in this pathology; as a result of increased inflammatory activity fibrinogen levels increase and thrombi form29,30(Figure 3).
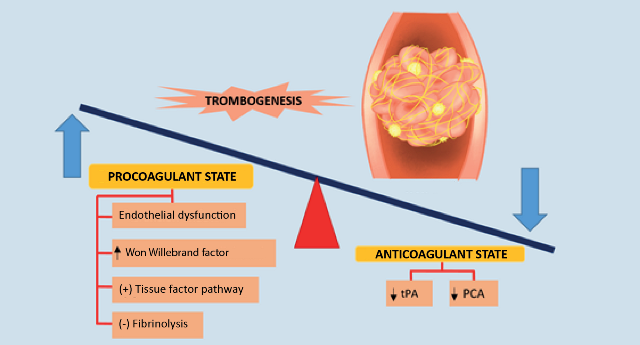
Increased levels of fibrin degradation products such as D-diameter and prolonged prothrombin time have been associated with poor prognosis in patients affected by the new betacoronavirus and multiple pathogenic mechanisms have been implicated in coagulopathy caused by COVID-19, including endothelial dysfunction, Von Willebrand factor elevation, Toll-like receptor activation and tissue factor pathway activation30.
The association with increased microvascular thrombosis, increased lactate dehydrogenase (LDH) and ferritin levels, and mild increases in prothrombin time (PT) and activated partial thrombopastin time (aPTT) are associated with thrombotic microangiopathy31.
The prevalence of arterial thrombosis is also high in patients with COVID-19 in whom the involvement of antiphospholipid antibodies has been suggested30 .
One study examined the presence of lower limb thrombosis without symptoms of deep vein thrombosis (TVP) by ultrasonography in patients with COVID-19 pneumonia treated in the intensive care unit (UCI) and reported a prevalence of 25%; this could be favoured by the time spent in bed, the described procoagulant state occurring in patients with COVID-19 and could also be due to a state of fibrinolytic inhibition32. The presence of deep vein thrombosis (DVT) is a recognised risk factor for developing PTE.
Due to the existing coagulopathy in patients with COVID-19, there are currently guidelines that recommend the use of anticoagulation in patients with COVID-19; there are even studies that show reduced mortality in cases with coagulopathy and treated with heparin (low molecular weight heparin) compared to patients who did have coagulopathy and were not treated with heparin33,34. However, treatment in relation to the coagulation alterations that occur in this disease is beyond the scope of this article.
SARS-CoV-2 and renal disorders
After pulmonary infection, the virus can enter the blood, accumulate in the kidneys and damage resident renal cells35. Among patients with COVID-19 and ARDS, a significant number of patients have been found to have acute renal dysfunction, some of whom even progressed to renal failure and required dialysis36.
Acute kidney injury (IRA) is a serious complication in critically ill patients, often leading to an increased risk of death37.
Possible pathogenic mechanisms of SDRA associated with IRA include haemodynamic alterations, including right heart failure, fluid overload and systemic congestion (including congestion of the renal vasculature), detrimental mechanical ventilation strategies, the development of secondary infections, immune-mediated inflammatory reaction with the release of high levels of harmful circulating inflammatory mediators that are capable of interacting with resident cells in the kidneys and causing endothelial dysfunction, microcirculatory disruption and tubular injury; virus-mediated renal injury in the glomerulus and tubular system has also been postulated 36,38.
A study by Pan et al. showed relatively high co-expression of ACE2 receptor and the serine protease TMPRSS in podocytes and renal glomerulus proximal rectal tubule cells, which were identified as candidate host cells in IRA38.
Thus, prerenal IRA has been suggested, but glomerulonephritis and even certain forms of acute tubular necrosis can also be found, suggesting components of intrarenal IRA in these patients39. Clinically, patients may present with proteinuria (probably due to podocyte damage), haematuria, oliguria and increased serum creatinine levels36. Another study by Hirsch et al. described possible predictors of IRA in COVID-19, citing advanced age, male sex, diabetes mellitus, hypertension, history of cardiovascular disease, increased body mass index (BMI), mechanical ventilation, vasopressor drugs and a history of treatment with renal angiotensin-aldosterone inhibitor drugs39.
Recently, a case of COVID-19 associated with collapsing glomerulopathy has also been described, which corresponds to the most feared variant of focal segmental glomerulonephritis. This pathology has been commonly associated with viral infections such as human immunodeficiency virus, parvovirus, cytomegalovirus, autoimmune diseases, heroin consumption and a greater predisposition in african americans40.
SARS-CoV-2 and gastrointestinal disturbances
Diarrhea is a common symptom in coronavirus infections; it has been detected in up to 30% of patients with MERS and 10.6% of patients with SARS-CoV-1941.
The ACE2 receptor required for viral entry was also expressed in upper esophageal cells as well as in ileum and colon enterocytes; TMPRSS2 is also co-expressed at the level of enterocytes and in the esophagus, which would explain the presence of gastrointestinal manifestations in patients with COVID-1912. In the pathogenesis of SARS-CoV-2 diarrhoeal syndrome, it is postulated that the viral infection causes an alteration of intestinal permeability, resulting in malabsorption by enterocytes42. The ACE2 receptor is required for surface expression of small intestinal amino acid transporters and it has been suggested that virus activity may cause enzymatic modifications, increasing susceptibility to intestinal inflammation and diarrhea; since amino acids such as tryptophan regulate the secretion of antimicrobial peptides by Paneth cells through activation of the mTOR pathway, these antimicrobial peptides could affect the composition and diversity of the microbiota contributing to pathogenesis leading to enteritis (inflammation of the small intestine) and ultimately diarrhea12.
SARS-CoV-2 and neurological disorders
Characteristic neurological manifestations have been described in some patients with COVID-19, indicating that SARS-CoV-2 may represent an underestimated opportunistic pathogen of the brain43.
In fact, several techniques such as immunohistochemistry, electron microscopy and polymerase chain reaction (PCR) have been used and the presence of coronavirus in the brain has been detected44,45.
Possible routes of entry of SARS-CoV-2 into the central nervous system (CNS) are postulated as trans-synaptic transfer in infected neurons, via the olfactory nerve, the lymphatic pathway and through the blood-brain barrier46.
It is believed that there is a retrograde axonal transport of SARS-CoV-2, where axonal microtubules are used to facilitate the transport of the virus into the CNS; the route of entry in this case is thought to be the olfactory neurons, as a study has been described with mice in which chemical ablation of these neurons protected them from infection47 (Figure 4).
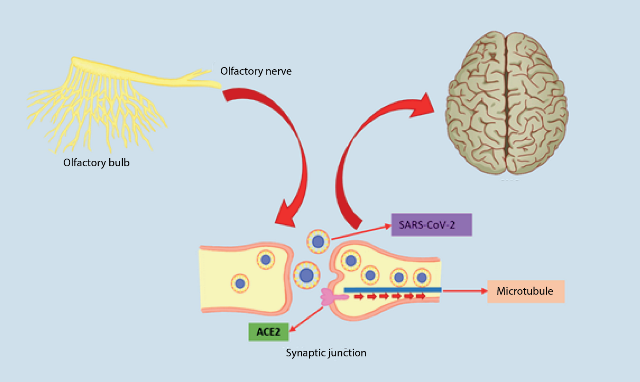
In the event that SARS-CoV-2 enters the CNS by crossing the blood-brain barrier (BHE), it could be thanks to a previous infection of the endothelial cells that form part of this barrier (along with astrocytes, pericytes and extracellular matrix) due to the large amount of ACE2 receptor that these cells contain; once it has reached this point, the virus would be able to invade adjacent tissue and cross the BHE, causing vascular and neuronal damage47 . Leukocytes could also carry the virus through the blood to the BHE, this mechanism of transport to the CNS would be facilitated by an increase in BHE permeability that occurs in the inflammation caused by SARS-CoV-2 infection, thus facilitating the passage of the virus46,48.
The most common neurological symptoms reported are headache, anosmia and ageusia and other neurological findings include cerebrovascular accident (ACV), impaired consciousness, coma, seizures and encephalopathy46. Toscano et al. reported cases of COVID-19 associated with Guillain-Barré syndrome (GBS), which presented 5-10 days after infection, GBS could be explained by molecular mimicry in which infectious viruses share epitopes similar to peripheral nerve components, which stimulates autoreactive T or B cells, a mechanism similar to that occurring with Campylobacter Jejuni infections and some viral infections such as Epstein-Barr virus, cytomegalovirus and Zika virus49.
SARS-CoV-2 and skin lesions
Cutaneous manifestations associated with COVID-19 have been reported in some case series. Patterns ranging from maculopapular erythema, urticaria, vesicular lesions, periflexural purpura, transient livedo reticularis, acroischemia, to erythema multiforme50, 51 have been described. The pathophysiological mechanisms of COVID-19 skin changes are still not well understood and it is not yet clear whether the skin changes are secondary to the respiratory infection or are instead a primary infection of the skin itself; it is postulated that viral particles present in the cutaneous blood vessels in patients with COVID-19 infection could lead to a lymphocytic vasculitis causing these changes52. This finding is also described in relation to a haematogenous spread of the virus through the cutaneous vascular system where there would also be an activation of the immune system with the mobilisation of lymphocytes and Langerhans cells; viral particles could induce the creation of immune complexes, leading T-helper cells (CD4) to produce cytokines such as IL-1, IFN-γ and TNF-α and to recruit eosinophils, cytotoxic T-cells (CD8), B-cells and natural killer (NK) cells leading to lymphocytic thrombophilic arteritis53.
Currently, while there are case series reports and articles referring to COVID-19-associated dermal lesions, further studies are needed to assess whether these lesions are directly associated with the virus, or attributable to the inflammatory response caused by the virus, and the details of the pathological mechanisms involved.
SARS-CoV-2 and cardiac disorders
Babapoor-Farrokhran et al. have suggested that underlying cardiovascular disease may predispose patients to an increased risk of coronavirus infection through increased systemic inflammation and immune system dysregulation; however, there is no scientific evidence to support this claim54.
There is now a growing literature exploring the cardiac lesions that may be associated with COVID-19. It is known that the ACE2 receptor is expressed in cardiac tissue specifically at the pericyte level, and in a parallel study by Burrel et al. it has been shown that the ACE2 receptor is expressed to a greater extent in patients with underlying cardiac pathology than in people with normal cardiac tissue55,56.
Patients receiving drugs such as angiotensin receptor antagonists (ARBs) or angiotensin II-converting enzyme inhibitors (ACE inhibitors) have a positive up-regulation of ACE2, therefore this receptor would be available in large quantities to provide a site
binding site for SARS-CoV-257. Cardiac manifestations of COVID-19 have been linked to an adrenergic response, systemic inflammatory state, direct viral infection of endothelial and myocardial cells, hypoxia due to respiratory failure, electrolyte imbalances, fluid overload and side effects of certain drugs directed against COVID-1957, 58, 59.
Zou et al. in a meta-analysis study examined the correlation of certain laboratory parameters and cardiac injury in patients hospitalised with COVID-19, finding significantly higher CRP, procalcitonin and NT-proBNP values59. There is a case reported in April 2020, which describes the direct involvement of coronavirus particles in the myocardium, causing a “fulminant myocarditis” with a significant increase in troponin and cardiac enzyme levels, progressing to cardiogenic shock that would have caused the collapse of the heart patient60.
Cardiac arrhythmias in these patients have been attributed to several factors, including myocardial inflammation caused by viral infection leading to electrophysiological and structural remodelling; but arrhythmias may also be secondary to medication side effects; atrial fibrillation was the most commonly observed cardiac arrhythmia in patients with COVID-1961 infection. There are reports of ventricular arrhythmias and torsada de pointes as complications of adverse effects due to drugs that prolong the QT interval on the electrocardiogram, especially azithromycin and hydroxychloroquine61, 62.
Patients with a medical history of coronary heart disease are prone to COVID-19 induced heart disease, and patients may develop acute coronary syndrome mainly as a thrombotic complication63.
Conclusions
SARS-CoV-2 is a new betacoronavirus that is considered an emerging virus in the global population. Understanding the structure and replication of the virus is essential to understand the pathological aspects of the virus. To date, SARS-CoV-2 has infected millions of people and affected billions of lives. The understanding of the disease and its pathophysiology in patients with COVID-19 is evolving, and clinicians must continue to conduct research, not only to understand the effects of the infection on different tissues and organs, but also to understand the possible short- and long-term complications of the disease, knowledge of which could also facilitate the management and treatment of these patients.
Potencial conflict of interest: he authors declare that they have no conflict of interest relevant to the submission of this article.
References
1. Gandhi RT, Lynch JB, Del Rio C. Mild or moderate Covid-19. N Engl J Med. 2020 April 24. DOI: 10.1056/NEJMcp2009249 [E-pub ahead of print]. https://doi.org/10.1056/NEJMcp2009249 [ Links ]
2. Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol. 2020; 92: 418-23. https://doi.org/10.1002/jmv.26234 [ Links ]
3. Cui J, Li F , Shi Z L. Origin and evolution of pathogenic coronaviruses. Microbiology. 2019; 17(3): 81-192. https://doi.org/10.1038/s41579-018-0118-9 [ Links ]
4. Renu K, Prasanna P L , Gopalakrishnan A V. Coronaviruses pathogenesis, comorbidities and multi-organ damage - A review, Life Sci. 2020 May 22. https://doi.org/10.1016/j.lfs.2020.117839 [E-pub ahead of print]. https://doi.org/10.1016/j.lfs.2020.117839 [ Links ]
5. Berlin D A, Gulick R M, Martinez F J. Severe Covid-19 . N Engl J Med. 2020 May 15. DOI: 10.1056/NEJMcp2009575 [E-pub ahead of print]. https://doi.org/10.1056/NEJMcp2009575 [ Links ]
6. Bhatraju P K, Ghassemieh B J, Nichols M, Kim R, Jerome K R, Nalla A K, et al. Covid-19 in critically ill patiente in the Seattle region - case series. N Engl J Med. 2020; 382(21): 2012-22. https://doi.org/10.1056/NEJMoa2004500 [ Links ]
7. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T., et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020; 5(7): 1-8. https://doi.org/10.1001/jamacardio.2020.1017 [ Links ]
8. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497-506. https://doi.org/10.1016/S0140-6736(20)30183-5 [ Links ]
9. Murray P R, Rosenthal K S, Pfaller M A Microbiología médica. 8va ed. Barcelona: Elsevier; 2017.p.506-511.
10. Fehr A.R, Perlman S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol Biol. 2015; 1282:1-23. https://doi.org/10.1007/978-1-4939-2438-7_1 [ Links ]
11. Vellingiri B., Jayaramayya K., Iyer M., Narayanasamy A., Govindasamy V., Giridharan B., et al. COVID-19: A promising cure for the global panic. Sci Total Environ. 2020; 725:1-18. https://doi.org/10.1016/j.scitotenv.2020.138277 [ Links ]
12. D’Amico F., Baumgart D.C., Danese S., Peyrin Biroulet L. Diarrhea During COVID-19 Infection: Pathogenesis, Epidemiology, Prevention, and Management. Clin Gastroenterol Hepatol.2020; 18(8): 1663-72. https://doi.org/10.1016/j.cgh.2020.04.001
13. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020; 181(2): 271-80. https://doi.org/10.1016/j.cell.2020.02.052 [ Links ]
14. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah N G, Decroly E. The spike glucoprotein of the new coronavirus 2019-CoV containsa furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020; 176: 1-5. https://doi.org/10.1016/j.antiviral.2020.104742 [ Links ]
15. Kim Y., Jedrzejczak R, Maltseva N I , Wilamowski M, Endres M, Godzik A,et al. Crystal structure of Nsp15 endoribonucleasa NendoU from SARS-CoV-2. Protein Sci. 2020; 29:1596-1605. https://doi.org/10.1002/pro.3873 [ Links ]
16. Oliva Marín J.E. SARS-CoV-2 origen, estructura, replicación y patogénesis. Alerta. 2020; 3(2):79-86. https://doi.org/10.5377/alerta.v3i2.9619 [ Links ]
17. Frieman M., Baric R. Mechanisms of Severe Acute Respiratory Syndrome Pathogenesis and Innate Immunomodulation. Microbiol Mol Biol Rev. 2008; 72(4): 672-85. https://doi.org/10.1128/MMBR.00015-08 [ Links ]
18. Giwa A, Desai A., Duca A. Novel 2019 Coronavirus SARS-CoV-2 (COVID-19): An Overview for Emergency Clinicians. Emerg Med Pract. 2020; 22(5): 1-24. [ Links ]
19. Blanco-Melo D., Nilsson-Payant B.E, Liu W.C, Uhl S., Hoagland D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19, Cell. 2020; 181 (5): 1036-45. https://doi.org/10.1016/j.cell.2020.04.026
20. Janeway C.A., Jr., Medzhitov R. Innate Immune Recognition. Annu Rev Immunol. 2002; 20 : 197-216. https://doi.org/10.1146/annurev.immunol.20.083001.084359
21. Lazear H.M., Schoggins J.W., Diamond M S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity. 2019; 50(4): 907-923. [ Links ]
22. Chu H., Chan J. F-W., Wang Y., Yuen T. T-T., Chai Y., Hou Y., et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis. 2020 April 9. DOI: 10.1093/cid/ciaa410. ciaa4. [E-pub ahead of print]. https://doi.org/10.1093/cid/ciaa410 [ Links ]
23. Li H., Liu L., Zhang D., Xu J., Dai H., Tang N., Su X., et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. 2020; 395: 1517-20. https://doi.org/10.1016/S0140-6736(20)30920-X [ Links ]
24. Xu Z., Shi L., Wang Y., et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020; 8: 420-22 . https://doi.org/10.1016/S2213-2600(20)30076-X [ Links ]
25. Grossman S.C, Porth C.M. Fisiopatología. Alteraciones en la salud. Conceptos básicos. 9na ed. España : Wolters Kluver; 2014. p. 988-92.
26. Iba T., Levy J.H., Levi M., Connors J.M., Thachil J. Coagulopathy of Coronavirus Disease 2019. Crit Care Med. 2020 May 26; Doi: 10.1097 / CCM.0000000000004458 [E-pub ahead of print]. https://doi.org/10.1097/CCM.0000000000004458 [ Links ]
27. López-Salvio Y.M., Herrera-Rodríguez L.J., Guzmán-Silahua S., Nava-Zavala A.H., Rubio-Jurado B. Dímero D: papel en patología trombótica. El Residente. 2018; 13(1): 12-22. [ Links ]
28. Klok F.A., Kruip M.J.H.A., van der Meer N.J.M., Arbous M.S., Gommers D.A.M.P.J., Kant K.M.,et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thrombosis Research. 2020; 191: 145-47. https://doi.org/10.1016/j.thromres.2020.04.041 [ Links ]
29. Delabranche X., Helms J., Meziani F. Immunohaemostasis: a new view on haemostasis during sepsis. Ann Intensive Care. 2017; 7(1): 117. [ Links ]
30. Giannis D., Ziogas I.A., Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS CoV-1, MERS-CoV and lessons from the past. J Clin Virol. 2020 June. https://doi.org/10.1016/j.jcv.2020.104362 [E-pub ahead of print]. https://doi.org/10.1016/j.jcv.2020.104362 [ Links ]
31. VCampbell C.M., Kahwash R. Will Complement Inhibition Be the New Target in Treating COVID-19-Related Systemic Thrombosis?. Circulation. 2020; 141(22): 1739-41. https://doi.org/10.1161/CIRCULATIONAHA.120.047419 [ Links ]
32. Danzi G.B., Loffi M., Galeazzi G., Gherbesi E. Acute pulmonary embolism and COVID-19 pneumonia: a random association?. Eur Heart J. 2020; 41(19): 1858. https://doi.org/10.1093/eurheartj/ehaa254 [ Links ]
33. Thachil J., Tang N., Gando S., Falanga A., Cattaneo M., Levi M., Clark C., et al. ISTH Interim Guidance on Recognition and Management of Coagulopathy in COVID-19. J Thromb Haemost. 2020; 18(5): 1023-26. https://doi.org/10.1111/jth.14810 [ Links ]
34. Thachil J. The versatile heparin in COVID-19. J Thromb Haemost. 2020; 18:1020-22. https://doi.org/10.1111/jth.14821 [ Links ]
35. Cheng Y., Luo R., Wang K., Zhang M., Wang Z., Dong L., et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020; 97: 829-38. https://doi.org/10.1016/j.kint.2020.03.005 [ Links ]
36. Fanelli V., Fiorentino M., Cantaluppi V., Gesualdo L., Stallone G., Ronco C., et al. Acute kidney injury in SARS-CoV-2 infected patients. Crit Care. 2020; 24: 155. https://doi.org/10.1186/s13054-020-02872-z [ Links ]
37. Wilson J.G., Calfee C.S. ARDS Subphenotypes: Understanding a Heterogeneous Syndrome. Crit Care. 2020; 24: 102. https://doi.org/10.1186/s13054-020-2778-x [ Links ]
38. Xiu-wu P., Da X., Hao Z., Wang Z., Lin hui W., Xin gang C.. Identifcation of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med. 2020 March 31; DOI: 10.1007/s00134-020-06026-1 [E-pub ahead of print]. https://doi.org/10.1007/s00134-020-06026-1 [ Links ]
39. Hirsch J.S., Ng J.H., Ross D.W., Sharma P., Shah H.H., Barnett R.L., et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020; 98(1): 509-12. https://doi.org/10.1016/j.kint.2020.05.006 [ Links ]
40. Larsen C.P., Bourne T.D., Wilson J.D., Saqqa O., Sharshir M.A. Collapsing Glomerulopathy in a Patient With Coronavirus Disease 2019 (COVID-19). Kidney Int Rep. 2020; 5: 935-39. https://doi.org/10.1016/j.ekir.2020.04.002 [ Links ]
41. Chan J. F-W., Yuan S., Kok K-H., Kai-Wang K., Chu H., et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020; 395 (10223): 514-523. https://doi.org/10.1016/S0140-6736(20)30154-9 [ Links ]
42. Gu J., Han B., Wang J. COVID-19: Gastrointestinal Manifestations and Potential Fecal-Oral Transmission. Gastroenterology. 2020; 158(6): 1518-19. https://doi.org/10.1053/j.gastro.2020.02.054 [ Links ]
43. Li Z., Liu T., Yang N., Han D., Mi X., Li Y., et al. Neurological manifestations of patients with COVID-19: potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front Med. 2020 May 4. https://doi.org/10.1007/s11684-020-0786-5 [E-pub ahead of print]. https://doi.org/10.1007/s11684-020-0786-5 [ Links ]
44. Netland J., Meyerholz D.K., Moore S., Cassell M., Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol. 2008; 82(15): 7264-75. https://doi.org/10.1128/JVI.00737-08 [ Links ]
45. Iroegbu J.D., Ifenatuoha C.W., Ijomone O.M. Potential neurological impact of coronaviruses: implications for the novel SARS-CoV-2. Neurol Sci. 2020 May 18. https://doi.org/10.1007/s10072-020-04469-4 [E-pub ahead of print]. https://doi.org/10.1007/s10072-020-04469-4 [ Links ]
46. Zubair A.S., McAlpine L.S., Gardin T., et. al. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019. JAMA Neurol. 2020; 77 (8): 1018-27. https://doi.org/10.1001/jamaneurol.2020.2065 [ Links ]
47. Baig A.M., Khaleeq A., Ali U., Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020; 11(7):995-98. https://doi.org/10.1021/acschemneuro.0c00122 [ Links ]
48. Sankowski R., Mader S., Valdés-Ferrer S.I. Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci. 2015; 9(28): 28. https://doi.org/10.3389/fncel.2015.00028 [ Links ]
49. Toscano G., Palmerini F., Ravaglia S., et al. Síndrome de Guillain-Barré asociado con SARS-CoV-2. N Engl J Med. 2020; 382 (26): 2574-76. https://doi.org/10.1056/NEJMc2009191 [ Links ]
50. Galván Casas C., Català A., Carretero Hernández G., Rodríguez-Jiménez P., Fernández Nieto D., Rodríguez-Villa Lario A., et al. Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol. 2020; 183(1): 71-7. https://doi.org/10.1111/bjd.19163 [ Links ]
51. Marzano A.V., Cassano N., Genovese G., Moltrasio C., Vena G.A. Cutaneous manifestations in patients with COVID-19: A preliminary review of an emerging issue. Br J Dermatol. 2020 June 1; DOI: 10.1111/BJD.19264. . [E-pub ahead of print]. https://doi.org/10.1111/bjd.19264 [ Links ]
52. Sachdeva M., Gianotti R., Shah M., Lucia B., Tosi D., Veraldi S., et al. Cutaneous manifestations of COVID-19: Report of three cases and a review of literature. J Dermatol Sci. 2020; 98(2): 75-81. https://doi.org/10.1016/j.jdermsci.2020.04.011 [ Links ]
53. Gianotti R., Zerbi P., Dodiuk-Gad R.P. Clinical and Histopathological study of skin dermatoses in patients affected by COVID-19 infection in the Northern part of Italy. J Dermatol Sci. 2020; 98(2): 141-3. https://doi.org/10.1016/j.jdermsci.2020.04.007 [ Links ]
54. Babapoor-Farrokhran S., Gill D., Walker J., et al. Myocardial injury and COVID-19: Possible mechanisms. Life Sci. 2020 April 28; DOI: https://doi.org/10.1016/j.lfs.2020.117723 [E-pub ahead of print]. https://doi.org/10.1016/j.lfs.2020.117723 [ Links ]
55. Burrel L.M., Risvanis J., Kubota E., Dean R.G., MacDonald P.S., Lu S., et al. Myocardial infartion increases ACE2 expression in rat and humans. Eur Heart J. 2020; 26(4): 369-75. https://doi.org/10.1093/eurheartj/ehi114 [ Links ]
56. Dong M., Zhang J., Ma X., Tan J., Chen L., Xin Y., Zhuang L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed Pharmacother. 2020 August 24; DOI: https://doi.org/10.1016/j.biopha.2020.110678 [E-pub ahead of print]. https://doi.org/10.1016/j.biopha.2020.110678
57. Boukhris M., Hillani A., Azzalini L. Cardiovascular Implications of the COVID-19 Pandemic: A global Perspective. Can J Cardiol. 2020; 36(7): 1068-80. https://doi.org/10.1016/j.cjca.2020.05.018 [ Links ]
58. Graham Atkinson J. Problems with the analysis in “Treatment with Hydroxychloroquine, Azithromycin, and Combination in Patients Hospotalized with COVID-19”. Int J Infect Dis. 2020; 99:37. https://doi.org/10.1016/j.ijid.2020.07.057
59. Zou F., Qian Z., Wang Y., Zhao Y., Bai J. CJC Open. 2020 June 23; DOI: https://doi.org/10.1016/ j.cjco.2020.06.010. [E-pub ahead of print]. [ Links ]
60. Tavazzi G., Pellegrini C., Maurelli M., Belliato M., Sciutti F., Bottazzi A., et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020; 22(5): 911-15. https://doi.org/10.1002/ejhf.1828 [ Links ]
61. Babapoor-Farrokhran S., Tarighati R., Gill D., Babapoor S., Amanullah A. Arrhythmia in COVID-19. Sn Compr Clin Med. 2020 August 14; DOI: 10.1002/ejhf.1828 [E-pub ahead of print]. https://doi.org/10.1002/ejhf.1828 [ Links ]
62. Villamañán E., Armada E., Ruano M. Drug-induced QT interval prolongation: Do we know the risks?. Clinic Med. 2020; 144(6): 269-74. https://doi.org/10.1016/j.medcli.2014.01.027 [ Links ]
63. Sánchez-Recalde A., Solano-López J.,Miguelena-Hycka J., Martín-Pinacho J.J., Sanmartín M., Zamorano J.L. COVID-19 and cardiogenic shock. Different cardiovascular presentations with high mortality. Rev Esp Cadiol. 2020; 73(8): 669-72. https://doi.org/10.1016/j.recesp.2020.04.018. [ Links ]


