Introducción
El síndrome de la espalda inclinada (SEI) también conocido como camptocormia (del griego kamptós: inclinado y kormós: tronco) se caracteriza por una flexión involuntaria de la columna toracolumbar durante la bipedestación, que se reduce en decúbito. Es una anomalía postural adquirida cuyas causas son diferentes de las de las cifosis estructurales. El SEI también se diferencia del síndrome de la Torre de Pisa (pleurothotonus), habitualmente caracterizado por una flexión lateral del tronco secundaria a una distonía tardía por el uso prolongado de neurolépticos.1
El SEI fue descrito por Henry Earle, en 1815, y Brodie, en 1818, y reportado por James Parkinson en alguno de sus casos, en 1817.2 El término “camptocormia” fue descrito, en 1816, por los neurólogos franceses Souques y Rosanoff, para indicar una incurvación del tronco secundaria a una “flexión histérica” en los soldados de la I Guerra Mundial que habían sufrido un shock durante la batalla.3 Durante un siglo, la camptocormia fue considerada una condición psiquiátrica hasta que Kiuru y Laroche4,5 fueron los primeros en asociarla a enfermedades orgánicas. Actualmente las causas del SEI son muy numerosas, inclusive secundarias a anomalías genéticas, como en el caso de la distrofia miotónica tipo 2 con una mutación del gen ZNF95 o en la atrofia multisistémica.6
El objetivo de este artículo es describir las causas del SEI y cómo abordar su tratamiento.
Presentamos cuatro casos clínicos de SEI de origen muscular: tres por atrofia aislada de la musculatura paravertebral espinal y uno, por miopatía inflamatoria; en este caso, el paciente había sido intervenido en tres ocasiones y los síntomas de SEI habían empeorado.
Además de la clínica de SEI, todos también sufrían dolor lumbar por el que habían recibido tratamiento mediante rizólisis, infiltraciones facetarias y fisioterapia, con escasa mejoría. A los fines diagnósticos, a todos se les realizaron una resonancia magnética (RM) de cuerpo entero para buscar compromiso de otros grupos musculares, análisis de laboratorio para detectar enfermedad inflamatoria o autoinmune y una biopsia muscular (Tabla 1).
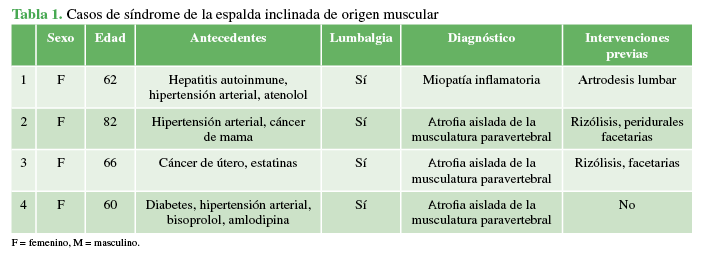 Tabla 1
Casos de síndrome de la espalda inclinada de origen muscular
Propia
Tabla 1
Casos de síndrome de la espalda inclinada de origen muscular
Propia
Caso 1
Mujer de
63 años que consulta por una cirugía fallida de columna. Había sido operada de
la columna lumbar, en tres ocasiones, en los últimos dos años, sin resolución
de los síntomas: dolor lumbar e inclinación anterior del tronco. Presenta una
cifosis toracolumbar reductible, con incapacidad de deambular en posición
erguida (Figura 1). Actualmente sin instrumentación lumbar (última cirugía de
retirada) (Figura 2). Camina asistida por caminador y, para mantener la
posición erecta, se empuja de los muslos (gesto similar a la maniobra de Gowers
de los pacientes con distrofia muscular de Duchenne). En la RM realizada antes
de la primera cirugía, se observa una columna lumbar sana, con discretos signos
de espondilosis, pero con una musculatura paravertebral erectora lumbar con
sustitución grasa (Figura 3). La RM muscular de cuerpo entero revela compromiso
de varios grupos musculares de forma simétrica. En la biopsia de la musculatura
paravertebral y gemelo, se identifican fibras necróticas entre la musculatura,
con inflamación intersticial; en el estudio inmunohistoquímico, se destaca la
sobrexpresión del MHC tipo I en las membranas de células musculares, con
abundantes macrófagos en perimisio y endomisio. El diagnóstico es miopatía
inflamatoria. A los pocos meses, tiene una hepatitis autoinmune, por lo que
recibe tratamiento con prednisona y azatioprina; mejoran los valores de las
enzimas hepáticas, pero no el SEI, ya que, en la musculatura paravertebral, los
cambios de sustitución grasa ya estaban instaurados.
 Figura 1
Figura 1
Paciente con incapacidad para mantener la postura erecta en bipedestación, reductible en decúbito o con el apoyo, lo que demuestra un desequilibrio en el plano sagital no estructurado.
Propia
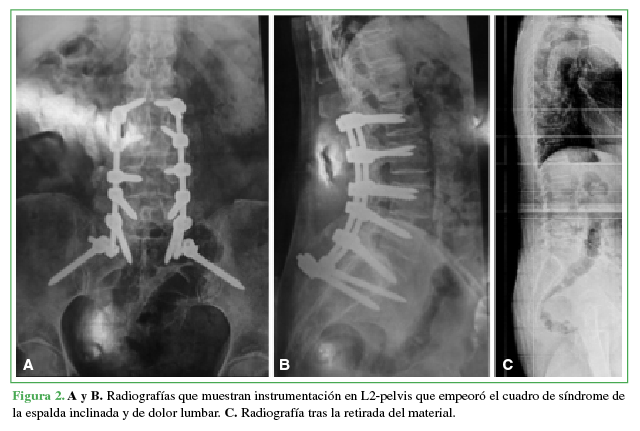 Figura 2
A y B.Radiografías que muestran instrumentación en L2-pelvis que empeoró el cuadro de síndrome de la espalda inclinada y de dolor lumbar. C. Radiografía tras la retirada del instrumental.
Propia
Figura 2
A y B.Radiografías que muestran instrumentación en L2-pelvis que empeoró el cuadro de síndrome de la espalda inclinada y de dolor lumbar. C. Radiografía tras la retirada del instrumental.
Propia
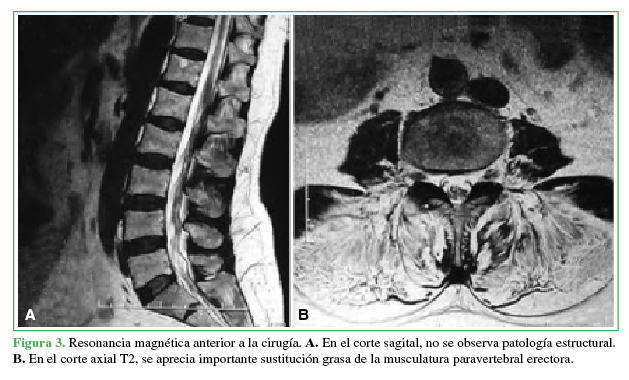 Figura 3
Resonancia magnética anterior a la cirugía.
Figura 3
Resonancia magnética anterior a la cirugía.
A. En el corte sagital no se observa patología estructural. B. En el corte axial T2 se aprecia imortante sustitución grasa de la musculatura paraespinal erectora
Propia
Casos 2, 3 y 4
Los tres
pacientes consultaron por dolor lumbar progresivo al mantener la postura
erecta. Todos tenían diferentes grados de inclinación anterior del tronco, que
se reducía en decúbito, sin patología estructural de columna (Figuras 4 y 5).
Ninguno tenía antecedentes de miopatías ni de enfermedad de Parkinson. A todos
se les solicitaron análisis de laboratorio, RM de cuerpo entero,
electromiografía y se les diagnosticó atrofia aislada de la musculatura
espinal. En la Tabla 1, se muestran los antecedentes patológicos de cada
paciente que se podrían relacionar con el desarrollo del SEI, aunque su
vinculación directa no pudo ser demostrada (síndromes paraneoplásicos, uso de
estatinas, diabetes mellitus). A todos ellos se les desaconsejó la cirugía y
fueron tratados mediante ortesis toracolumbar o lumbosacra, fisioterapia y el
uso complemetario de andadores como asistentes durante la marcha.
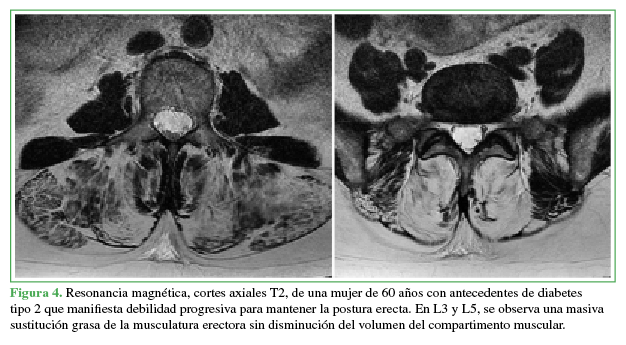 Figura 4
Resonancia magnética de cortes axiales T2 de una mujer de 60 años con antecedentes de diabetes tipo 2 que manifiesta debilidad progresiva para mantener la posición erecta.
Figura 4
Resonancia magnética de cortes axiales T2 de una mujer de 60 años con antecedentes de diabetes tipo 2 que manifiesta debilidad progresiva para mantener la posición erecta.
En L3 y L5 se observa una masiva sustitución grasa de la musculatura erectora sin disminución del volumen del compartimiento muscular
Propia
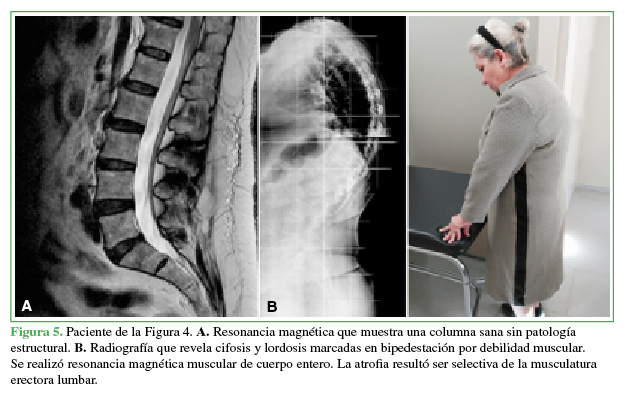 Figura 5
Paciente de la figura 4
Figura 5
Paciente de la figura 4
A. Resonancia magnética que muestra una columna sana sin patología estructural. B. Radiografía que revela cifosis y lordosis marcadas en bipedestación por debilidad muscular. Se realizó resonancia magnética muscular de cuerpo entero. La atrofia resultó ser selectiva de la musculatura erectora lumbar.
Discusión
Presentación clínica
En la
actualidad, no hay criterios bien definidos para caracterizar el SEI. La
mayoría de los estudios utilizan el ángulo de inclinación anterior del tronco
de entre 15° y 45° como criterio mayor debido a una debilidad progresiva de la
musculatura paravertebral extensora.7-9 El SEI se manifiesta
progresivamente en bipedestación o al deambular y mejora con el decúbito.10
En algunos casos, es indoloro, aunque en la mayoría, se asocia con dolor lumbar
axial, sobre todo cuando se asocia con la enfermedad de Parkinson.11-13
El SEI difícilmente se confunde con el “síndrome de la cabeza caída” (dropped head syndrome), aunque ambos
cuadros comparten muchas de las causas que los originan, se presentan juntos en
muy raras ocasiones.14 Se estima que la edad media de presentación
clínica del SEI es de 65 años, el 69% de los pacientes tiene antecedentes de
enfermedad de Parkinson y el 25%, de distonía. Hasta el 50% tiene antecedentes
familiares de enfermedad muscular.7,12 Actualmente el SEI se
manifiesta, con más frecuencia, a causa de enfermedades orgánicas y solo, rara
vez, es secundario a una enfermedad psiquiátrica.15 La incidencia
general del SEI es desconocida. Solo se publicaron datos sobre su incidencia
para los trastornos del movimiento. La frecuencia relativa de las miopatías
primarias o secundarias es muy variable. En una cohorte de SEI de un centro de
reumatología, el 65% sufría una distrofia muscular; el 17%, enfermedad de
Parkinson; y el 13%, miositis.16 En una cohorte de un centro de
enfermedades neuromusculares, el 25% tenía distrofia escápulo-humeral y el 18%,
miositis por cuerpos de inclusión.14 Hay cierta evidencia de que la
distrofia escápulo-humeral está subestimada en el SEI.15
No existe
un consenso o una clasificación oficial sobre el SEI. Los neurólogos prefieren
dividir su etiología en las originadas por alteraciones del sistema nervioso
central o periférico; por el contrario, nosotros preferimos dividirlas en: de
origen muscular o de origen neurológico, según su respuesta al tratamiento
local o sistémico.7
SEI de origen muscular
En este subgrupo de pacientes, el SEI se origina por debilidad de la musculatura erectora paravertebral espinal.
La insuficiencia de estos músculos genera un desequilibrio postural por el cual el tronco se flexiona hacia adelante.
Al no ser una deformidad estructurada, la postura se corrige simplemente en decúbito.
La debilidad de la musculatura extensora puede ser un cuadro primario e idiopático, o secundario a varias enfermedades que generan cambios patológicos en toda la musculatura responsable de la extensión del tronco (Tabla 2). Los SEI de origen muscular pueden ser identificados por los cambios electromiográficos, hipodensidad muscular en la RM y cambios miopáticos en la biopsia muscular.
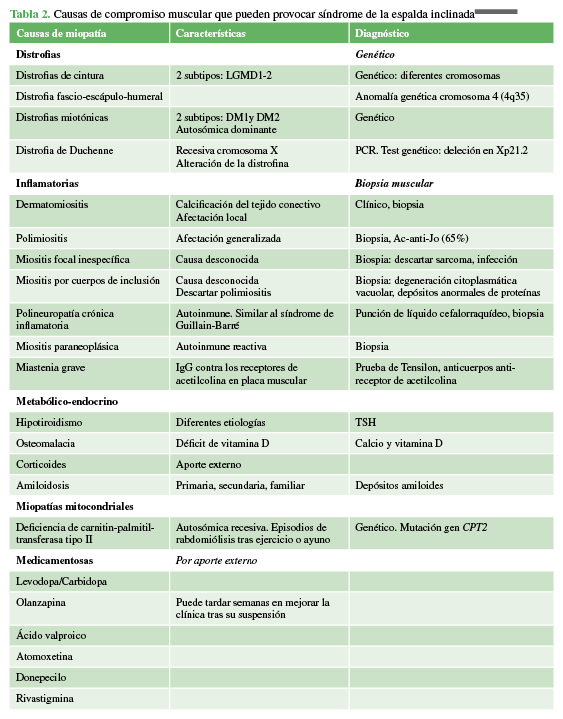 Tabla 2.
Causas de compromiso muscular que pueden provocar un síndrome de espalda inclinada. 14,19,22-24,38
Propia
Tabla 2.
Causas de compromiso muscular que pueden provocar un síndrome de espalda inclinada. 14,19,22-24,38
Propia
Muscular primario
- Miopatía primaria idiopática aislada de la musculatura
paravertebral
Es una de
las causas más frecuentes en la práctica clínica. Se manifiesta en adultos sin
antecedentes de enfermedades neurológicas o autoinmunes, que consultan por la
dificultad de mantener la posición erecta y una progresiva fatiga en el
transcurso del día. Los principales hallazgos son la infiltración grasa de la
musculatura paravertebral lumbar en los estudios por imágenes y en la biopsia.16,17
En 1991, Laroche y cols. publicaron, por primera vez, una serie de 16 pacientes
(media de la edad, 66 años) que se caracterizaba por la aparición tardía de una
miopatía limitada a la musculatura paravertebral. Todos tenían en común la
flexión anterior del tronco reductible en decúbito, infiltración grasa en la
musculatura paravertebral, según la tomografía computarizada (TC), un patrón
común en la biopsia y antecedentes familiares. Los autores postularon que esta
cifosis lumbar adquirida se debía a una miopatía primaria de inicio tardío,
localizada exclusivamente en la musculatura extensora paravertebral. Desde esta
primera publicación, esta hipótesis inicial fue respaldada por otros
investigadores17-19 y por posteriores estudios del mismo grupo.16
Otros autores confirman su etiología idiopática, con un predominio en el sexo
femenino y con algunos casos de herencia familiar.17,19,20La
debilidad muscular está estrictamente localizada en la musculatura extensora
paravertebral, sin otros déficits musculares clínicamente detectables. El SEI
es responsable de discapacidad y dolor lumbar importantes que empeoran con el
tiempo y tienen poca respuesta al tratamiento médico y rehabilitador.20
La herramienta diagnóstica más útil son los estudios por imágenes (TC o RM) de
las secciones lumbar y dorsal de la musculatura paravertebral.17 La
imagen típica es la musculatura paravertebral que conserva el volumen normal, pero
con completa sustitución grasa (Figuras 4 y 5), a diferencia de las de
etiología neurogénica por atrofia, en las que el volumen del músculo disminuye,
pero la densidad del tejido muscular es normal.21 En un estudio de
Ricq y Laroche, en el que se repite la TC a 23 pacientes a los 3.5 años, se
comunica que algunos también mostraban el mismo patrón de infiltración grasa en
los músculos glúteo medio y supraespinoso, muslos y pantorrillas,20
y que, en la electromiografía, se pueden detectar patrones miopáticos en el
glúteo medio y en el deltoides,22 aunque sin compromiso clínico en
estos grupos musculares. En todos los estudios, la anatomía patológica de las
biopsias musculares muestra un grado variable de reemplazo de las fibras
musculares por tejido adiposo y fibrosis.17,19 El SEI de origen
primario idiopático puede estar incluido en el grupo de distrofias musculares
progresivas de inicio tardío. Si bien varias investigaciones han encontrado una
herencia familiar, faltan estudios genéticos y moleculares para caracterizar el
modo de herencia, tal vez, se trate de una enfermedad congénita.23
Tomando todos estos datos se podría especular que esta miopatía de aparición
tardía y restringida a la musculatura axial podría estar vinculada a una de las
principales distrofias musculares, posiblemente a uno de los subtipos de
distrofias musculares de las cinturas.
Muscular secundario
El SEI
puede ser un síntoma de varias enfermedades musculares sistémicas. Las
distrofias son enfermedades genéticas hereditarias. La distrofia muscular
fascio-escápulo-humeral es un cuadro que provoca debilidad muscular progresiva
y que afecta principalmente a los músculos de la cara, los hombros y los
brazos. La anomalía genética está situada en la extremidad del brazo largo del
cromosoma 4 humano (4q35). Es uno de los muchos tipos de distrofias musculares
que existen y probablemente la distrofia que más se asocie al SEI23 y, en
algunos casos, es poco reconocida.15 Otras distrofias responsables
del SEI son las distrofias de cinturas y la distrofia miotónica de Steinert.23
El SEI puede ser secundario a miositis, como la dermatomiositis o polimiositis,
o la miositis focal o miositis por cuerpos de inclusión. Se puede diagnosticar
por electromiografía, valores elevados de creatincinasa en sangre y biopsia
muscular, cuya característica son los infiltrados perivasculares y endomisiales
inflamatorios con necrosis de las fibras musculares.24 Ciertos
trastornos metabólicos o endocrinos, como el hipotiroidismo y la osteomalacia o
la miopatía por corticoides, pueden producir un SEI secundario.18
Algunos medicamentos relacionados con el SEI, a los que también se los
considera un factor desencadenante del síndrome de Pisa, son el ácido
valproico, los agentes anticolinérgicos y los agonistas dopaminérgicos.25
Por último, se han publicado dos causas excepcionales de SEI: la miopatía
amiloide y la miopatía mitocondrial.26
SEI de origen neurológico
Entre las enfermedades neurodegenerativas, el SEI es más frecuente en pacientes con enfermedad de Parkinson.7 En estos casos, su prevalencia es del 3% al 17,6%, según los diferentes autores,11,26 y estudios de series más grandes informan una prevalencia del 10%.27 Los factores que predisponen al SEI en pacientes con enfermedad de Parkinson son: edad avanzada, sexo masculino, compromiso temprano de la musculatura axial (distonía axial), gravedad del Parkinson y una latencia de aparición >5 años del diagnóstico del Parkinson.28,29
Estudios recientes demuestran que la mayoría de los SEI neurológicos son consecuencia de lesiones en los ganglios basales, responsables de coordinar los reflejos posturales en flexión-extensión que hacen posible mantener la postura erecta.30 Esta función está mediada por la dopamina, producida, en parte, por la sustancia negra. El rol de la dopamina es fundamental en la función de los ganglios basales, y su alteración provoca cuadros, como la enfermedad de Parkinson y los síndromes Parkinson plus que incluyen la atrofia multisistémica y la parálisis supranuclear.27
Una de las características de la enfermedad de Parkinson en bipedestación es la discreta actitud en flexión del tronco hacia adelante relacionada con la distonía axial del parkinsonismo.31 En el SEI, la flexión anterior es severa y, a veces, se relaciona con escoliosis. El SEI en pacientes con enfermedad de Parkinson fue definido como una flexión anterior del tronco >45°.7,32 Se suele manifestar en un subgrupo de pacientes con Parkinson, en el cual la distonía axial es el síntoma predominante, con activación excesiva de la musculatura abdominal. El tratamiento con levodopa no tiene efecto en el SEI; sin embargo, mejora la acinesia, el temblor y la rigidez.7,23,32 La falta de efecto de la levodopa sobre el SEI ha sugerido que podría tratarse de una forma específica de parkinsonismo en el que otras disfunciones no dopaminérgicas de los ganglios basales podrían ser las responsables.23
Durante años, la distonía axial fue considerada la causa del SEI en los enfermos de Parkinson.33-35 En los últimos tiempos, se han descrito cambios focales miopáticos en la musculatura paraespinal de algunos pacientes con Parkinson, mediante electromiografía, TC, RM y biopsia, aunque su prevalencia no está bien documentada y el papel exacto de estos cambios y el desarrollo de SEI son desconocidos.32,36 Una nueva teoría sobre la desregulación de la capacidad de propiocepción explicaría el mecanismo del SEI en los pacientes con Parkinson. Los cambios miopáticos son iguales a los de un músculo que ha sufrido una tenotomía, en donde la tensión muscular es baja y el músculo recibe un estímulo de hipercontractilidad. A diferencia de una tenotomía en donde los cambios miopatológicos se deben a una desregulación del arco reflejo polisináptico a nivel de los tendones, los cambios miopatológicos en el SEI del Parkinson podrían obedecer a una desregulación de la propiocepción a nivel del sistema nervioso central. Este concepto fisiopatológico es mantenido debido a que las lesiones lenticulares pueden causar el SEI y que la neuroestimulación del núcleo subtalámico puede aliviar el SEI en enfermos con Parkinson, ya que devuelve parcialmente esa capacidad propioceptiva. Durante la evolución del SEI, cuando el músculo es remplazado por cambios crónicos de fibrosis y sustitución grasa, la neuroestimulación del núcleo subtalámico ya no tendría efecto en el músculo y, en consecuencia, tampoco en la inclinación del tronco.37
Aparte de la enfermedad de Parkinson, el SEI también puede aparecer en pacientes con atrofia multisistémica37,38 y demencia de cuerpos de Lewy,39 esclerosis lateral amiotrófica, sobre todo la que afecta inicialmente la musculatura respiratoria.40 En cuanto a los cuadros neurodegenerativos, se han descrito casos aislados de SEI en pacientes con enfermedad de Alzheimer.30
Diagnóstico
Como la
etiología del SEI es bastante heterogénea, proponemos un algoritmo diagnóstico
basado en datos clínico-familiares, análisis de sangre, estudios por imágenes
(TC, RM), electromiografía, biopsia muscular y, por último, estudio genético.
Examen clínico
Interrogar sobre antecedentes de enfermedades de componente genético y familiar, como las distrofias musculares, Parkinson (familias con alta incidencia de mutación de genes SPARK) o enfermedades metabólicas o autoinmunes.
Al principio, debe centrarse en los signos primarios de miopatías o trastornos del movimiento, como la enfermedad de Parkinson o las distonías. En el caso de las miositis, el músculo afectado puede ser asimétrico y tener signos inflamatorios. Se debe medir el grado de la inclinación del tronco con goniómetro, ya que esta puede variar con el tiempo y verificar que la inclinación no sea fija.41 La evolución del SEI puede también orientar hacia su etiología: las inflamatorias muestran un curso más rápido de semanas, en tanto que las primarias miopáticas progresan lentamente, en meses o años. Es importante documentar aspectos, como patologías de columna, consumo de medicación (neurolépticos), dolor lumbar y uso de soportes de la marcha (bastón, caminador, carrito de la compra, etc.).
Análisis de sangre
Hay una serie de parámetros bioquímicos que pueden orientar sobre la etiología del SEI, entre ellos, los reactantes de fase aguda (proteína C reactiva, eritrosedimentación) y la creatincinasa para las inflamatorias o autoinmunes, el metabolismo fosfocálcico, vitamina D, hormonas tiroideas, lactato y piruvato para las endocrino-metabólicas.7,12,23
Estudio neurofisiológico
La electromiografía es una herramienta muy útil para el diagnóstico del SEI. Según su causa, la electromiografía puede ser normal, neurogénica o miopática. La elección de los grupos musculares por estudiar dependerá del examen físico. Puede comprender la musculatura dorsal y ventral del tronco (paravertebrales y los rectos abdominales) en pacientes con trastornos del movimiento. Cuando se sospecha una miopatía se deberá estudiar también la musculatura proximal y distal de los miembros.
En los ancianos, el patrón neurogénico y la actividad espontánea no son necesariamente patológicos.42 Las unidades motoras de la musculatura paravertebral difieren de la de los miembros en que estas tienen menor amplitud y duración.19 Un patrón miogénico también puede detectarse en los pacientes con enfermedad de Parkinson.8
Diagnóstico por imágenes
La TC o
la RM cerebrales pueden mostrar signos de atrofia, calcificaciones de los
ganglios basales, lesiones lenticulares o anomalías del tronco.11 En
los casos de distrofia o de miopatía selectiva idiopática, la RM de la
musculatura paravertebral puede detectar rasgos de una miopatía circunscrita,
como grados variables de atrofia y de sustitución grasa de la musculatura
erectora (Figura 4).43 Los cambios agudos musculares que produzcan
edema se pueden observar en la secuencia STIR de la RM, como en el caso de las
miositis. En pacientes con enfermedad de Parkinson, los hallazgos pueden variar
desde cambios agudos con edema y tumefacción de la musculatura paravertebral,
el cuadrado lumbar y el psoas, en los primeros 31 meses,36,44 hasta
los más habituales hallazgos tardíos crónicos de degeneración y sustitución
grasa.18,45,46 Una desventaja de la RM muscular es su falta de
especificidad. Ante el hallazgo de hiperintensidades en el STIR muscular, se
debería indicar una biopsia muscular con el objetivo de diferenciar la miositis
de un trauma, desnervación u otras causas de enfermedades musculares.45
Biopsia muscular
La indicación más frecuente de biopsia muscular en el SEI sirve para identificar o excluir miositis. La biopsia de la musculatura paraespinal no se puede remplazar tomando una muestra de cualquier otro grupo muscular; por ello, se aconseja la guía previa mediante estudios por imágenes (RM, TC, ecografía). Se debe tener precaución para no tomar la muestra en el mismo sitio donde se realizó la electromiografía, ya que los trayectos de la aguja podrían imitar cambios inflamatorios. En la Tabla 2, se enumera el espectro de cuadros por considerar.
En pacientes con enfermedad de Parkinson, la biopsia muscular muestra un patrón constante compuesto por cambios miopáticos con hipertrofia de las fibras tipo 1, pérdida de las fibras tipo 2, pérdida de la actividad oxidativa enzimática y una fina capa granular de la ácido-fosfatasa reactiva a las lesiones.44 También se observa una variable extensión de fibrosis del endomisio y degeneración grasa que parece correlacionarse con la duración del SEI.32
Tratamiento
El
tratamiento del SEI dependerá principalmente de su etiología.
Tratamiento del SEI en
pacientes con enfermedad de Parkinson
En la mayoría de los pacientes, la levodopa no mejora la clínica de SEI,7,32 a excepción de los pacientes con distonía on-off y los que tienen fluctuaciones de fin de dosis.46 Optimizar la terapia de la enfermedad de Parkinson puede mejorar el SEI, como se describió con la infusión continua de apomorfina a cinco pacientes con un seguimiento de tres años.47
Algunos pacientes con distonía de la musculatura abdominal podrían beneficiarse parcialmente de la inyección de toxina botulínica en los músculos rectos abdominales, oblicuo externo y psoas ilíaco.7,48,49
Se ha demostrado que la neuroestimulación bilateral del núcleo subtalámico mejora los principales síntomas del Parkinson.46,50 Sin embargo, la neuroestimulación ha demostrado ser eficaz en el SEI secundario al Parkinson, si se efectúa dentro de los primeros 1.5 años del inicio de los síntomas de la inclinación del tronco. Entre los 17 meses y los 3 años, los resultados son ambiguos y, a partir de los 40 meses, ningún paciente ha mejorado, posiblemente porque, en la musculatura, ya se han instaurado los cambios crónicos atróficos y de sustitución grasa.37,51
Tratamiento quirúrgico del
SEI en pacientes con Parkinson
La
mayoría de los casos publicados de SEI tratados con cirugía son de pacientes
con enfermedad de Parkinson. La alta tasa de complicaciones en estos pacientes
parecería estar relacionada con su alta susceptibilidad a la osteoporosis.33
Los pacientes con Parkinson son una población de alto riesgo quirúrgico, tienen
una tasa de complicaciones médicas del 30%, de complicaciones quirúrgicas del
52% y una tasa de revisión quirúrgica del 35%.34 Los procedimientos
quirúrgicos sobre la columna en pacientes con Parkinson aumentan el riesgo de
desarrollar un SEI, inclusive el riesgo de producir una inestabilidad, sobre
todo, a nivel adyacente, es más alto en los procedimientos menores.9
Tratamiento del SEI de origen
distónico
Hay algunos casos publicados que han mejorado con tratamiento médico único de levodopa50 o en combinación con tetrabenazina, pimozida o agentes anticolinérgicos.51 Los pacientes con una distonía aislada de la musculatura abdominal podrían beneficiarse con las inyecciones de toxina botulínica en los rectos abdominales y en el oblicuo externo.49 Como última alternativa, la estimulación cerebral profunda del globo pálido interno resultó eficaz en este subgrupo de pacientes con SEI por una distonía primaria.52
Tratamiento del SEI en
pacientes con miopatías inflamatorias y miopatías
primarias
En el caso de las miopatías inflamatorias o las polineuropatías desmielinizantes, la elección de tratamiento inmunosupresor dependerá del cuadro específico y de su estadio.
Para los pacientes con miositis focal, el tratamiento con corticoides por vía endovenosa será suficiente.45,53En casos secundarios a polimiositis o dermatomiositis, la combinación de corticoides e inmunosupresores (metotrexato, azatioprina, ciclosporina) deberá estar sujeta a la actividad de la propia enfermedad.10 Una excepción es la miositis por cuerpos de inclusión, para la cual el tratamiento inmunosupresor y las inmunoglobulinas son ineficaces, por lo que el enfoque terapéutico se deberá centrar en la fisioterapia.54
Tratamiento del SEI con
ortesis
Independientemente
de la causa del SEI, los pacientes se pueden beneficiar de una ortesis para
corregir la inclinación del tronco y el dolor lumbar. En un estudio prospectivo
de Sèze y cols. en el que se utilizó una ortesis de distracción anterior
toracopelviana, el SEI tuvo una mejoría radiográfica y clínica significativa.55
Los caminadores con ruedas o el apoyo en el carro de la compra pueden ser un
material de soporte efectivo para mejorar la habilidad para caminar y reducir
también el dolor lumbar.56,57
Conclusión
El
pronóstico del SEI, cualquiera sea su causa, es pobre, los síntomas suelen
progresar hasta afectar la bipedestación de manera irreversible.
Bibliografía
1. Yeh YW, Chen YC, Chen CK, Feng HM, Wang TY, Chen CL. Chronic Pisa syndrome associated with switching antipsychotics from olanzapine to ziprasidone. Prog Neuropsychopharmacol Biol Psychiatry 2009;33(1):162-3. https://doi.org/10.1016/j.pnpbp.2008.11.001
2. Parkinson J. An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 2002;14(2):223-36. https://doi.org/10.1176/jnp.14.2.223
3. Souques A. Réforme, incapacité, gratifications dans la camptocormie. Rev Neurol 1916;23:757-8.
4. Laroche M, Rousseau H, Mazieres B, Bonafe A, Joffre F, Arlet J. Value of densitometry in muscular pathology. Personal cases and review of the literature. Rev Rhum Mal Osteoartic 1989;56:433-9.
5. Serratrice G. Clinical semiology of neuromuscular diseases. Bent spine myopathy or syndrome. Acta Myol 2007;26:1-4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2949321/
6. Reichel G, Kirchhöfer U, Stenner A. Camptocormia-segmental dystonia. Proposal of a new definition for an old disease. Nervenarzt 2001;72:281-5. https://doi.org/10.1007/s001150050751
7. Azher SN, Jankovic J. Camptocormia: pathogenesis, classification and response to therapy. Neurology 2005;65:355-9. https://doi.org/10.1212/01.wnl.0000171857.09079.9f
8. Margraf NG, Wrede A, Rohr A, Schulz-Shaeffer WJ, Raethjen J, Eymess A, et al. Camptocormia in idiopathic Parkinson´s disease: a focal myopathy of the paravertebral muscles. Mov Disord 2010;25(5):542-51. https://doi.org/10.1002/mds.22780
9. Nakane S, Yoshioka M, Oda N, Tani T, Cida K, Suzuki M, et al. The characteristics of camptocormia in patients with Parkinson´s disease: a large cross-sectional multicenter study in Japan. J Neurol Sci 2015;358(1-2):299-303. https://doi.org/10.1016/j.jns.2015.09.015
10. Kuo SH, Vullanganti M, Jimenez-Shahed J, Kwan JY. Camptocormia as a presentation of generalized inflammatory myopathy. Mucle Nerve 2009;40(6):1059-63. https://doi.org/10.1002/mus.21403
11. Lepoutre AC, Devos D, Blanchard-Dauphin A, Pardessus V, Maurage CA, Ferriby D, et al. A specific clinical pattern of camptocornia in Parkinson´s disease. J Neurol Neurosurg Psychiatry 2006;77(11):1229-34. https://doi.org/10.1136/jnnp.2005.083998
12. Ricq G, Laroche M. Acquired lumbar kyphosis caused in adults by primary paraspinal myopathy. Epidemiology, computed tomography findings and outcomes in a cohort of 23 patients. Joint Bone Spine 2000;67(6):528-32. https://doi.org/10.1016/S1297-319X(00)00203-7
13. Margraf NG, Granert O, Hampel J, Wrede A, Schulz-Schaeffer WJ, Deuschl G. Clinical definition of camptocormia in Parkinson’s disease. Mov Disord Clin Pract 2016;4(3):349-57. https://doi.org/10.1002/mdc3.12437
14. Ghosh PS, Milone M. Camptocormia as presenting manifestation of a spectrum of myopathic disorders. Muscle Nerve 2015;52(6):1008-12. https://doi.org/10.1002/mus.24689
15. Skidmore F, Anderson K, Fram D, Weiner W. Psychogenic camptocormia. Mov Disord 2007;22(13):1974-5. https://doi.org/10.1002/mds.21708
16. Laroche M, Cintas P. Bent spine syndrome (camptocornia): a retrospective study of 63 patients. Join Bone Spine 2010;77(6):593-6. https://doi.org/10.1016/j.jbspin.2010.05.012
17. Doherty KM, Noyce AJ, Silveira-Moriyama L, Nisbet A, Quinn N, Lees AJ. Familiar camptocornia: from dystonia to myopathy. J Neurol Neurosurg Psychiatry 2012;83(3):350-1. https://doi.org/10.1136/jnnp.2011.246561
18. Laroche M, Ricq G, Delise MB. Bent spine syndrome: computed tomography study and isokinetic evaluation. Muscle Nerve 2002;25(2):189-93. https://doi.org/10.1002/mus.10016
19. Mahjneh I, Marconi G, Paetau A, Saarinen A, Salmi T, Somer H. Axial myopathy: an unrecognised entity. J Neurol 2002;249(6):730-4. https://doi.org/10.1007/s00415-002-0701-9
20. Ricq G, Laroche M. Cyphose lombaire acquise de l ádulte par myopathie primitive des muscles paravertébraux. Aspects épidémiologiques, tomodensitométriques et évolutifs, à propos d´une cohorte de 23 patients. Rev Rhum 2000;67(10):908-13. https://doi.org/10.1016/S1169-8330(00)00034-X
21. Bulke JA, Crolla D, Termote JL, et al. Computed tomography of muscle. Muscle Nerve 1981;4(1):67-72. https://doi.org/10.1002/mus.880040112
22. Laroche M, Delisle MB, Aziza R, Lagarrigue J, Mazieres B. Is camptocormia a primary muscular disease? Spine 1995;20(9):1011-6. https://doi.org/10.1097/00007632-199505000-00007
23. Emery AE. The muscular dystrophies. Lancet 2002;359(9307): 687-95. https://doi.org/10.1016/S0140-6736(02)07815-7
24. Hund E, Heckl R, Goebel HH, Meinck HM. Inclusion body miositis presenting with isolated erected spinae paresis. Neurology 1995;45(5):993-4. https://doi.org/10.1212/wnl.45.5.993
25. Suzuki T, Matsuzaka H. Drug-induced Pisa syndrome (pleurothotonus). Mol Diag Ther 2002;16(3):165. https://doi.org/10.2165/00023210-200216030-00003
26. Delcey V, Hachulla E, Michon-Pasturel U, Queyrel V, Hatron PY, Boutry N, et al. La camptocormie: un signe de myopathie axiale. À propos de sept observationes. Rev Med Interne 2002;23(2):144-54. https://doi.org/10.1016/S0248-8663(01)00530-6
27. Yorikata A, Shimo Y, Takanashi M, Fukae J, Hatano T, Nakahara T, et al. Motor and non-motor symptoms of 1453 patients with Parkinson’s disease: prevalence and risks. Parkinsonism Relat Disord 2013;19(8):725-31. https://doi.org/10.1016/j.parkreldis.2013.04.001
28. Spuler S, Krug H, Klein C, Medialdea IC, Jacob W, Ebersbach G, et al. Myopathy causing camptocormia in idiopathic Parkinson’s disease: a multidisciplinary approach. Mov Disord 2010;25(5):552-9. https://doi.org/10.1002/mds.22913
29. Tiple D, Fabbrini G, Colosimo C, Ottaviani D, Camerota F, Defazio G, et al. Camptocormia in Parkinson disease. J Neurol Neurosurg Psychiatry 2009;80(2):145-8. https://doi.org/10.1136/jnnp.2008.150011
30. Dietz V, Zijlstra W, Assaiante Ch, Trippel M, Berger W. Balance control in Parkinson´s disease. Gait Posture 1993;1(2):77-84. https://doi.org/10.1016/0966-6362(93)90018-V
31. Jankovic J, Tintner R. Dystonia and parkinsonism. Parkinson Dis Relat Disord 2001;8(2):109-21. https://doi.org/10.1016/S1353-8020(01)00025-6
32. Bloch F, Houeto JL, Tezena du Montcel S, Bonneville F, Etchepare F, Welter ML, et al. Parkinson´s disease with camptocormia. J Neurol Neurosurg Psychiatry 2006;77(11):1223-8. https://doi.org/10.1136/jnnp.2006.087908
33. Babat L, McLain R, Bingaman W, Kalfas I, Young P, Rufo Smith C. Spinal surgery in patients with Parkinson’s disease: construct failure and progressive deformity. Spine (Phila Pa 1976) 2004;29(18):2006-12. https://doi.org/10.1097/01.brs.0000138306.02425.21
34. Koller H, Acosta F, Zenner J, Ferraris L, Hitzl W, Meier O, et al. Spinal surgery in patients with Parkinson’s disease: experiences with the challenges posed by sagittal imbalance and the Parkinson´s spine. Eur Spine J 2010; 19(20):1785-94. https://doi.org/10.1007/s00586-010-1405-y
35. Micheli F, Pardal M. Dopa-responsive dystonic camptocormia. Neurology 2007;68(18):1543. https://doi.org/10.1212/01.wnl.0000265316.61721.63
36. Wunderlich S, Csoti I, Reiners K,
Günthner-Lengsfeld T, Schneider C, Becker G, et al. Camptcormia in Parkinson’s
disease mimicked by focal myositis of the paraspinal muscles. Mov Disord 2002;17(3):598-600. https://doi.org/10.1002/mds.10110
37. Schulz-Schaeffer W, Margraf N, Munser S, Wrede A, Buhmann C, Deuschl G, Oehlwein C. Effect of neurostimulation on camptocormia in Parkinson’s disease depends on symptoms duration. Mov Disord 2015;30(3):368-72. https://doi.org/10.1002/mds.26081
38. Skidmore F, Mikolenko I, Weiss H, Weiner W. Camptocormia in patients with multiple system atrophy. Mov Disord 2005;20(8):1063-4. https://doi.org/10.1002/mds.20521
39. Gavrylova N, Limousin N, Belin J, Praline J. Camptocormia as presenting sign in dementia with Lewy bodies. Clin Neurol Neurosurg 2013;115(11):2397-8. https://doi.org/10.1016/j.clineuro.2013.09.005
40. Gautier G, Verschueren A, Monnier A, Attarian S, Salort-Campana E, Pouget J. ALS with respiratory onset: clinical features and onset of non-invasive ventilation on the prognosis. Amyotroph Lateral Scler 2010;11(4):379-82. https://doi.org/10.3109/17482960903426543
41. Melamed E, Djaldetti R. Camptocormia in Parkinson´s disease. J Neurol 2006;253(Suppl 7):VII14-6. https://doi.org/10.1007/s00415-006-7004-5
42. Barkhaus PE, Periquet MI,
Nandedkar SD. Quantitative motor unit action potential analysis in paraspinal
muscles. Muscle Nerve
1997;20(3):373-5. https://doi.org/10.1002/(SICI)1097-4598(199703)20:3<373::AID-MUS19>3.0.CO;2-1
43. Haig AJ, Tong HC, Kendall R. The bent spine syndrome: myopathy + biomechanics = symptoms. Spine J 2006; 6(2):190-4. https://doi.org/10.1016/j.spinee.2005.08.002
44. Margraf NG, Rohr A, Granert O, Hampel J, Drews A, Deuschl G. MRI of lumbar trunk muscles in patients with parkinson´s disease and camptocornia. J Neurol 2015;262(7):1655-64. https://doi.org/10.1007/s00415-015-7726-3
45. Ohana M, Durand MC, Marty C, Lazareth JP, Maisonobe T, Mompoint D, et al. Whole body muscular RMN to detect myopathies in non-extrapyramidal bent spine syndrome. Skeletal Radiol 2014;43:1113-22. https://doi.org/10.1007/s00256-014-1909-3
46. Yamada K, Shinojima N, Hamasaki T, Kuratsu J. Subthalamic nucleous stimulation improves Parkinson’s disease-associated camptocormia in parallel to its preoperative levodopa responsiveness. J Neurol Neurosurg Psychiatry 2016;87(7):703-9. https://doi.org/10.1136/jnnp-2015-310926
47. Mensikova K, Kaiserova M, Vastik M, Kurkova S, Kanovsky P. Treatment of camptocormia with continuous subcutaneous infusions of apomorphine: 1-year prospective pilot study. J Neural Transm (Vienna) 2015;122(6):835-9 https://doi.org/10.1007/s00702-014-1297-9. Erratum: J Neural Transm (Vienna) 2015;122(9):1353.https://doi.org/10.1007/s00702-015-1385-5
48. Kataoka H, Tonomura Y, Eura N, Terashima M, Kawahara M, Ueno S. Painful abdominal contractions in patients with Parkinson disease. J Clin Neurosci 2012;19(4):624-7. https://doi.org/10.1016/j.jocn.2011.06.026
49. Bertram KL, Stirpe P, Colosimo C. Treatment of camptocormia with botulinum toxin. Toxicon 2015;107(Pt A):148-53. https://doi.org/10.1016/j.toxicon.2015.06.004
50. Deuschl G, Schade-Brittinger C, Krack P, Volkmann J, Schafer H, Botzel K, et al. A randomized trial of deep-brain stimulation for Parkinson´s disease. N Engl J Med 2006;355(9):896-908. https://doi.org/10.1056/NEJMoa060281
51. Bathia K, Quinn N, Mardsen C. Clinical features and natural history of axial predominant adult onset primary dystonia. J Neurol Neurosurg Psychiatry 1997;63(6):778-91. https://doi.org/10.1136/jnnp.63.6.788
52. O’Riordan S, Paluzzi A, Liu X, Aziz T, Nandi D, Bain P. Response to bilateral globus pallidus internal stimulation in three patients. Mov Disord 2009;24(Suppl. 1):1-653 p489. https://doi.org/10.1002/mds.22628
53. Dominik J, Sheenan G, Schleimer J, Wixom C. Response to the dropped hear/bent spine syndrome to treatment with intravenous immunoglobulin. Muscle Nerve 2006;33(6):824-6. https://doi.org/10.1002/mus.20506
54. Benveniste O. Inclusion-body myositis. Rev Med Interne 2014;35(7):472-9. https://doi.org/10.1016/j.revmed.2013.09.001
55. de Sèze M, Creuze A, Mazaux J. An orthosis and physiotherapy programme for camptocormia: a prospective case study. J Rehabil Med 2008;40(9):761-5. https://doi.org/10.2340/16501977-0252
56. Capecci M, Serpicelli C, Fiorentini L, Censi G, Ferretti M, Orni C, et al. Postural rehabilitation and Kinesio taping for axial postural disorders in Parkinson´s disease. Arch Phys Med Rehabil 2014;95(6):1067-75. https://doi.org/10.1016/j.apmr.2014.01.020
57. Upadhyaya C, Starr P, Mummaneni P. Spinal deformity and Parkinson disease: a treatment algorithm. Neurosurg Focus 2010;28(3):e5. https://doi.org/10.3171/2010.1.FOCUS09288
Información adicional
Nivel de Evidencia:: IV
Conflicto de intereses: Los autores no declaran conflictos de intereses.
Cómo citar este
artículo: Covaro A, Vilà-Canet G, Ciccolo F, Rodriguez-Alabau S, Garcia de Frutos
A, Ubierna-Garcés M, Isart Torruela A, Cancer Castillo D, Cáceres-Palou E.
Síndrome de la espalda inclinada.
Presentación de cuatro casos y revisión de la bibliografía. Rev Asoc Argent Ortop Traumatol
2019;84(4):393-405. http://dx.doi.org/10.15417/issn.1852-7434.2019.84.4.924




