
Presentación de caso
Síndrome de activación macrofágica en paciente pediátrico con enfermedad de Still
Macrophage activation syndrome in a pediatric patient suffering from Still's disease
Universidad Médica Pinareña
Facultad de Ciencias Médicas de Pinar del Rio Dr. Ernesto Ché Guevara de la Serna, Cuba
ISSN: 1990-7990
Periodicidad: Frecuencia continua
vol. 15, núm. 1, 2019
Recepción: 12 Noviembre 2018
Aprobación: 22 Diciembre 2018
Publicación: 01 Enero 2019

Resumen: Introducción: la enfermedad de Still en la infancia constituye un trastorno inflamatorio, de origen autoinmune, que se enmarca dentro del grupo de las artritis idiopáticas juveniles, constituyendo probablemente la entidad más peculiar del grupo. Suele cursar en forma de brotes de actividad repetidos, intercalados por períodos de remisión. Presentación del caso: se presenta el caso de un paciente de dieciséis años de edad, con antecedentes de artritis juvenil sistémica, con un cuadro clínico de 72 horas de evolución caracterizado por fiebre intermitente, tos, piodermitis, exantema generalizado y artritis. Durante la evolución clínica se consideró activación de la enfermedad de base versus sepsis con base en los criterios clínicos, lo cual fue sustentado posteriormente por los hallazgos de laboratorio. Recibió tratamiento con antibióticos de amplio espectro, antiinflamatorios no esteroideos, esteroides a razón de 1mg/kg/día, sin presentar mejoría. Posteriormente, se inició tratamiento con pulsos de metilprednisolona e inmunomoduladores. Finalmente, se diagnosticó un síndrome de activación macrofágica, con base en los parámetros de laboratorio clínico. Evolucionó tórpidamente y falleció en la Unidad de Cuidados Intensivos a los 22 días de su ingreso. Conclusiones: el síndrome de activación macrofágica constituye una de las formas secundarias dentro del grupo de linfohistiocitosis hemocitofágicas. Son muy diversos los factores que en este contexto se han asociado a su aparición. Clínicamente estos procesos se caracterizan por fiebre prolongada que no cede a pesar de antibióticos. Característicamente la artritis suele estar ausente. La primera línea de tratamiento la constituyen los corticoides a dosis altas.
Palabras clave: Síndrome De Activación Macrofágica, Macrófagos, Artritis, Artritis Juvenil, Artritis Reumatoide.
Abstract: Introduction: Still's disease in childhood constitutes an inflammatory disorder, of autoimmune origin, which is framed within the group of juvenile idiopathic arthritis, probably being the most peculiar entity of the group. It usually occurs in the form of repeated outbreaks of activity, combined with periods of remission. Case report: a sixteen-year-old patient with a history of systemic juvenile arthritis is presented, with a clinical picture of 72 hours of evolution characterized by intermittent fever, cough, pyodermitis, generalized rash and arthritis. During the clinical evolution, it was considered base-disease activation versus sepsis supported on clinical criteria, which was later corroborated by laboratory findings. The patient underwent treatment with broad-spectrum antibiotics, non-steroidal anti-inflammatory drugs, steroids at a rate of 1mg / kg / day, without improvement. Subsequently, the treatment with pulses of methylprednisolone and immunomodulator-therapy was initiated. Finally, a macrophage activation syndrome was diagnosed, supported on clinical laboratory parameters. Patient’s evolvement was slowly and died in the Intensive Care Unit, 22 days after the admission. Conclusions: the macrophage activation syndrome is one of the secondary forms within the group of hemophagocytic lymphohistiocytosis. The factors that in this context have been associated with its onset are very diverse. Clinically, these processes are characterized by prolonged fever that does not disappear despite antibiotic-therapy. Typically, arthritis is usually absent. The first line of treatment is corticosteroids at high doses.
Keywords: Macrophage Activation Syndrome, Macrophages, Arthritis, Arthritis, Juvenile, Arthritis, Rheumatoid.
INTRODUCCIÓN
La artritis idiopática juvenil sistémica(AIJ) o enfermedad de Still supone aproximadamente el 10% de los casos de AIJ, aunque es el grupo en el que se encuentran los pacientes más graves y en el que adquieren especial relevancia las manifestaciones extraarticulares que, de hecho, son necesarias para establecer el diagnóstico(1).
Con igual incidencia en ambos sexos, el 66% de los casos debutan antes de los 5 años. Clínicamente los brotes cursan con artritis, criterio imprescindible pero no constante al inicio, e importante afectación sistémica: fiebre alta intermitente de al menos 2 semanas de duración (debiendo haberse documentado que es diaria durante al menos 3 días), exantema maculo-papular evanescente, que característicamente aparece y desaparece con la fiebre, sin dejar lesión residual, linfadenopatías generalizadas, hepatoesplenomegalia y serositis, la más frecuente es la afectación pericárdica Analíticamente destaca la elevación de reactantes de fase aguda, anemia, leucocitosis, trombocitosis e hipergammaglobulinemia. La presencia de Anticuerpos antinucleares (ANA) variando entre un 10 y un 30 %(1,2).
El síndrome de activación macrofágica (SAM) es una complicación grave que puede aparecer en este contexto, caracterizada por hiperactividad reticuloendotelial descontrolada con hiperactividad inflamatoria potencialmente letal(2).
PRESENTACIÓN DEL CASO
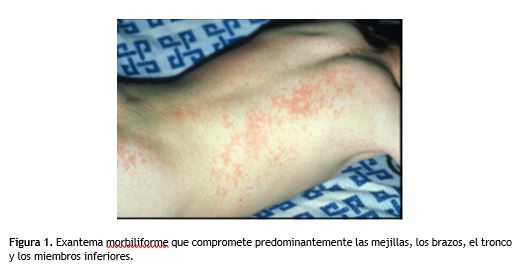
Paciente masculino de 16 años de edad, color de la piel blanca, diagnosticado a los 3 años de enfermedad de Still o artritis idiopática juvenil de inicio sistémico (AIJS). Su enfermedad clínicamente se ha caracterizado por exantema cutáneo asalmonado, de predominio troncular y porción proximal de extremidades, persistente (figura 1); episodios febriles, poliartritis, odinofagia, y pericarditis recurrentes que en una ocasión llegó al taponamiento cardíaco y requirió la realización de una ventana pericárdica; acompañado de elevación en plasma de leucocitos y reactantes de fase aguda (RFA), como la proteína C reactiva positiva.
Durante la evolución de su enfermedad ha presentado 10 brotes de actividad, tratados con indometacina, prednisona, metotrexate, cloroquina, azatioprina y salazosulfapiridina no llevando ninguno en los últimos 10 meses por permanecer asintomático. Dentro de los antecedentes patológicos familiares se recoge que la madre padece hipertensión arterial. El paciente nunca ha sido intervenido quirúrgicamente, ha recibido varias transfusiones de sangre en el curso de su enfermedad, no presenta hábitos tóxicos ni alergia medicamentosa.
Consultó por un cuadro clínico de 72 horas de evolución caracterizado por fiebre intermitente (de 38,5°C a 39°C), tos húmeda, piodermitis, exantema generalizado y artritis, con una Velocidad de Sedimentación Globular (VSG) de 133mm en 1ª hora y Reacción en cadena de la Polimerasa (PCR) de 95 mg/dL, leucocitosis y anemia ligera, sin otras alteraciones en la analítica. Se pautó prednisona 15 mg/día v.o. Ante la falta de mejoría, a los 3 días se incrementó la prednisona a 30 mg/día, sin mejorar por lo que acudió a Urgencias. En analítica se objetivó discreta alteración de enzimas hepáticas (ASAT 165 U/L y ALAT 112 U/L), con PCR similar. El estudio de sangre periférica, no objetivó blastos ni linfocitos activados y se informó como sugestivo de infección bacteriana.
La radiografía de tórax no presentaba alteraciones, fue diagnosticado de sepsis por piodermitis iniciándose tratamiento con antibiótico de amplio espectro. Tras 5 días sin presentar ninguna mejoría y con cultivos negativos, el paciente ingresó en la Unidad de Cuidados Intensivos, con un proceso infeccioso o un brote de actividad como principales diagnósticos diferenciales.
En la exploración a su ingreso el paciente estaba febril (Tª axilar de 38,8º C), presentando microadenopatías cervicales, auscultación cardiopulmonar normal, hepatomegalia de 3 cm y esplenomegalia de 4-5 cm, sinovitis, exantema generalizado, sin anomalías en exploración neurológica ni signos de irritación meníngea.
La analítica realizada mostró los siguientes resultados:
Leucocitos 6470/mm3 (Neutrófilos 1450, Linfocitos 4760)
Hemoglobina 8.6 gr/dL
Hematocrito 30.4 %
VCM 74.8 fL
Plaquetas 107000/mm3
PCR 2.53
VSG 131mm
Bilirrubina total de 1.71 mg/dL (directa = 1.2 mg/dL)
ASAT 562 U/L
ALAT 465 U/L
GGT 353 U/L
LDH 5550 mg/dL (R.N.240-480),
Triglicéridos 378 mg/dL
Albúmina 2.8 g/dl
Fibrinógeno 145 g/dl (200-450)
La ecografía abdominal confirmó la hepatoesplenomegalia, difusa, sin otras alteraciones. Otras pruebas realizadas (Rx Tórax, ecocardiografía, hemocultivos, uro y coprocultivo y cultivo de frotis faríngeo) fueron negativas.
Ante estos hallazgos se plantea la posibilidad de un síndrome de activación macrofágica, por lo que se inicia tratamiento intravenoso con metilprednisolona 2 mg/día en bolos y ciclofosfamida 0.75 mg/m2 de superficie corporal. Evolucionó tórpidamente y falleció en la Unidad de Cuidados Intensivos a los 22 días de su ingreso con hemorragia pulmonar y fallo multiórgano.
DISCUSIÓN
El SAM constituye una de las formas secundarias dentro del grupo de linfohistiocitosis hemocitofágicas, procesos caracterizados por infiltración multiorgánica de linfocitos T e histiocitos(3,4,5). En la etiopatogenia de estos procesos se implica el déficit de actividad citotóxica, algunos trabajos han atribuido dicha disfunción al déficit funcional y/o de síntesis de perforinas pudiendo asociar déficit poblacional y/o de actividad de células NK y linfocitos T CD 8, lo que permitiría un estímulo antigénico persistente con la consiguiente activación mantenida de la respuesta inmunológica, producción masiva y descontrolada de citoquinas e hiperproliferación celular(6). El SAM es una forma específica en el contexto de la enfermedad inflamatoria autoinmune, siendo su asociación más frecuente con la AIJIS, en algunas series se produce hasta en un 5-10 % de estos casos, pudiendo aparecer en el contexto de otros procesos tales como Still del adulto, LES, dermatomiositis, Kawasaki, etc. Son muy diversos los factores que en este contexto se han asociado a la aparición del SAM (infecciones bacterianas, fúngicas, víricas, parasitosis y un amplio grupo de fármacos) (7,8).
Clínicamente estos procesos se caracterizan por fiebre prolongada que no cede a pesar de antibióticos, que suele diferir de la fiebre habitual de la AIJIS en que no sigue el patrón habitual en picos, manifestaciones cutáneas (rash, paniculitis), síndrome hemorrágico (epistaxis, hemorragia digestiva, lesiones purpúricas, etc.), hepatoesplenomegalia que puede acompañarse de ictericia y ascitis; pancreatitis aguda, linfadenopatía generalizada, afectación del Sistema Nervioso Cefalea (cefalea, letargia, irritabilidad, convulsiones, pudiendo llegar al coma)(9).
Se describen como menos frecuentes la afectación pulmonar (infiltrados de naturaleza inflamatoria, derrame pleural, distrés respiratorio), afectación cardiaca (miocardiopatía dilatada, pericarditis) y la afectación renal (fracaso renal agudo oligoanúrico, sd. nefrótico)(10,11,12).
Característicamente la artritis suele estar ausente. Analíticamente se caracteriza por la presencia de hipoalbuminemia, hipertransaminemia e hiperbilirrubinemia, marcada elevación de ferritina, aumento de LDH, descenso de VSG (en relación con hipofibrinogenemia), citopenia (puede afectarse cualquiera de las tres series y puede darse cualquier combinación de éstas), alteraciones de coagulación (con descenso de todos los factores y del fibrinógeno, prolongación de TP, APTT y elevación de dímero-D y los productos de degradación de la fibrina, como reflejo de la coagulopatía que suele instaurarse ya desde las primeras horas de evolución), niveles extremadamente elevados de IL-2, IL-6, IL-10, IL-12, IL-18, TNF-alfa, IFN-gamma, GMCSF, entre otras citoquinas(13).
Desde el punto de vista diagnóstico se han propuesto una serie de criterios clínicos, analíticos y anatomopatológicos pendientes de validación, es un diagnóstico en ocasiones difícil dado que los síntomas pueden ser difícilmente distinguibles de los presentes en períodos de actividad de la enfermedad de Still, los datos analíticos pueden quedar parcialmente enmascarados por el proceso de base, la característica hemofagocitosis no se objetiva hasta en un 50 % de casos, y parámetros como la cuantificación de la actividad de las células NK y la determinación de sCD25 requieren técnicas escasamente disponibles(14,15).
La primera línea de tratamiento la constituyen los corticoides a dosis altas, sin embargo hay bastantes diferencias en la literatura en lo que las dosis administradas se refiere, desde dosis de metilprednisolona de 2 mg/kg/día hasta pulsos de 20-30 mg/kg/día o 1 gr/m2/día, durante 3-5 días, lo que dependerá de la gravedad del cuadro clínico(16). En formas corticorresistentes o graves se asocia ciclosporina, inicialmente IV en perfusión continua, en dosis de 2-5 mg/kg/día (máximo 7 mg/Kg/día), con posterior continuación oral(17). En casos refractarios se ha comunicado respuesta anecdótica a la administración de inmunoglobulinas IV, globulina antitimocítica, etopósido, plasmaféresis, así como a etanercept, infliximab y anakinra(18,19).
Con todo ello el síndrome de activación macrofágica constituye un cuadro potencialmente letal, mortalidad 8-22 % de casos, con una tasa de recurrencias que oscila de 1 a 16 %, en el que el principal determinante pronóstico es la rapidez en la instauración del tratamiento, por lo que el aspecto fundamental en el manejo de estos pacientes es la alerta permanente ante la presencia de manifestaciones sugestivas de esta complicación(20).
CONCLUSIONES
El SAM constituye una de las formas secundarias dentro del grupo de linfohistiocitosis hemocitofágicas. Son muy diversos los factores que en este contexto se han asociado a su aparición. Clínicamente estos procesos se caracterizan por fiebre prolongada que no cede a pesar de antibióticos. Característicamente la artritis suele estar ausente. La primera línea de tratamiento la constituyen los corticoides a dosis altas.
Referencias
1. Azaña JM, Torrelo A, Matito A. Actualización en mastocitosis. Parte 1: fisiopatología, clínica y diagnóstico. Actas Dermo-Sifiliográficas [Internet]. 2016 [citado 2018 Nov 10]; 107(1): 5-14. Disponible en: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0001731015004019?returnurl=null&referrer=null
2. Villalobos C, Vásquez E, Villalobos JC, Venegas O. Linfohistiocitosis hemofagocítica: Reporte de un caso. Revista ANACEM [Internet]. 2012 [citado 2018 Nov 10]; 6(3): 155-157. Disponible en: http://eds.a.ebscohost.com/eds/detail/detail?vid=0&sid=2c7e2737-5d4e-493e-9e93-428ab1da96d8%40sessionmgr4009&bdata=Jmxhbmc9ZXMmc2l0ZT1lZHMtbGl2ZQ%3d%3d#AN=88417412&db=asx
3. Sterba G, Sterba Y, Iglesias AG. Síndrome de activación macrofágica en adultos con enfermedad reumática. Revista Colombiana de Reumatología [Internet]. 2016 [citado 2018 Nov 15]; 23(2): 137-143. Disponible en: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0121812315001243?returnurl=null&referrer=null
4. Egües Dubuc C, Aldasoro Cáceres V, Uriarte Ecenarro M, Errazquin Aguirre N, Hernández Rubido I, et al. Síndrome de activación macrofágica secundario a enfermedades autoinmunes, hematológicas, infecciosas y oncológicas. Serie de 13 casos clínicos y una revisión bibliográfica. Reumatología Clínica [Internet]. 2015 [citado 2018 Nov 15]; 11(3): 139-143. Disponible en: https://www.sciencedirect.com/science/article/pii/S1699258X1400134X
5. Mahuad CV, Garate GM, Vicente Reparaz MA, Casali C, Del Olmo M, Bolgiani A. Secondary hemophagocytic syndrome due to recurrent infections in a severely burned patient. Medicina [Internet]. 2013 Jun [citado 2018 Oct 28]; 73(3): 255-258. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S0025-76802013000300010&lng=es
6. Clemente Garulo D, León Mateos L, López Robledillo JC. Enfermedades reumáticas en la adolescencia. Artritis ideopática juvenil. Conectivopatias. Medicine-Programa de Formación Médica Continuada Acreditado [Internet]. 2018 [citado 2018 Dic 10]; 12(61): 3588-3600. Disponible en: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0304541218301896?returnurl=null&referrer=null
7. Lequerré T, Vittecoq O. Pruebas de laboratorio en patología articular. Exploración práctica de la inmunidad innata y adaptativa (humoral y celular). EMC-Aparato Locomotor [Internet]. 2015 [citado 2018 Nov 11]; 48(2): 1-6. Disponible en: https://www.sciencedirect.com/science/article/pii/S1286935X15711123
8. Nieto-González JC, Monteagudo I. Estado actual del tratamiento con infiltraciones intraarticulares en la artritis ideopática juvenil. Reumatología Clínica [Internet]. 2018 [citado 2018 Dic 10]; en Prensa. Disponible en: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S1699258X18301748?returnurl=null&referrer=null
9. Brieva Herrero MT, Pérez I, Cardenas M, Isla B. Manejo de artritis idiopática juvenil sistémica en pediatría con agentes biológicos: a propósito de un caso. Farm Hosp. [Internet]. 2014 Ago [citado 2018 Oct 29]; 38(4): 384-385. Disponible en: http://dx.doi.org/10.7399/FH.2014.38.4.7368
10. Bautista Espinosa KA, Fossas Garciadiego P, Rodríguez león E. Síndrome hemofagocítico. Conceptos actuales. Gaceta Médica de México [Internet]. 2013 [citado 2018 Ago 25]; 149(4): 431-457. Disponible en: http://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=45536
11. Jacobson JL, Pham JT. Juvenile Idiopathic Arthritis: A focus on Pharmacologic Management. Journal of Pediatric Health Care [Internet]. 2018 [citado 2018 Nov 10]; 32(5): 515-528. Disponible en: https://www.clinicalkey.es/#!/content/journal/1-s2.0-S0891524517306533
12. Higgins GC. Complications of treatments for Pediatric Rheumatic Diseases. Pediatric Clinics of North America [Internet]. 2018 [citado 2018 Nov 10]; 65(4): 827-854. Disponible en: https://www.clinicalkey.es/#!/content/journal/1-s2.0-S0031395518300476
13. Díaz Martín D, Úbeda Cantera M, López Suárez A, Álvarez de Mon Soto M. Respuesta inmune innata y sus complicaciones fisiopatológicas. Medicine-Programa de Formación Médica Continuada Acreditado [Internet]. 2017 [citado 2018 Nov 12]; 12(24): 1388-1397. Disponible en: https://www.sciencedirect.com/science/article/pii/S0304541216302347
14. Mostaza-Fernández JL, Laso JG, Ule DC, Morales JR. Linfohistiocitosis hemofagocítica asociada a infecciones virales: reto diagnóstico y dilema terapéutico. Revista Clínica Española [Internet]. 2014 [citado 2018 Nov 15]; 214(6): 320-327. Disponible en: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0014256514001283?returnurl=null&referrer=null
15. León-Pedroza JI, González-Tapia LA, del Olmo-Gil E, Castellanos-Rodríguez D, Escobedo G, González-Chávez A. Inflamación sistémica de grado bajo y su relación con el desarrollo de enfermedades metabólicas: de la evidencia molecular a la aplicación clínica. Cirugía y Cirujanos [Internet]. 2015 [citado 2018 Nov 10]; 83(6): 543-551. Disponible en: https://www.sciencedirect.com/science/article/pii/S0009741115001188
16. Ravelli A, Daví S, Minoia F, Martini A, Cron RQ. Macrophage Activation Syndrome. Hematology/Oncology Clinics of North America [Internet]. 2015 [citado 2018 Nov 15]; 29(5): 927-941. Disponible en: https://www.clinicalkey.es/#!/content/journal/1-s2.0-S0889858815000878
17. Navarrete M, García-Sesnich J, Dutzan N, Henríquez L, Puente J, Gamonal J. Presencia de marcadores de las vías de activación de los macrófagos en periodontitis crónica. Rev. Clin. Periodoncia Implantol. Rehabil. Oral [Internet]. 2012 Dic [citado 2019 Ene 28]; 5(3): 131-134. Disponible en: https://scielo.conicyt.cl/scielo.php?script=sci_arttext&pid=S0719-01072012000300006&lng=es
18. Arreguin-Reyes R, Valle-Leal J, Lozano Rentería L, Medina-Valentón E, Álvarez Bastidas L. Descripción de una cohorte de pacientes de artritis idiopática juvenil en el estado de Sonora, México. Revista Colombiana de Reumatología [Internet]. 2016 [citado 2018 Oct 22]; 23(4): 236-241. Disponible en: https://www.clinicalkey.es/#!/content/journal/1-s2.0-S0121812316300421
19. Hernández-Rodríguez J, Ruis-Ortiz E, Yagüe J. Enfermedades autoinflamatorias monogénicas: conceptos generales y presentación en pacientes adultos. Medicina Clínica [Internet]. 2018 [citado 2018 Dic 23]; 150(2): 67-74. Disponible en: https://www.sciencedirect.com/science/article/pii/S0025775317306310
20. Cárdenas Bruno M, Moreno Miravalles MI. Diagnóstico posmortem de un caso con síndrome hemofagocítico secundario. Revista Cubana de Pediatría [revista en Internet]. 2018 [citado 2018 Nov 28]; 91(1): [aprox. 10 p.]. Disponible en: http://www.revpediatria.sld.cu/index.php/ped/article/view/490

