Resumen: Como una forma pedagógica de aprendizaje activo, se relacionaron temas desarrollados en clase de biología molecular con la pandemia del coronavirus COVID-19 que en este momento avanza en casi la totalidad del planeta. Contar con un método eficiente para diagnosticar la infección durante el inicio de esta infección respiratoria aguda es importante para detectar tanto los pacientes sintomáticos como los asintomáticos. Por este motivo, se retó a los estudiantes de séptimo semestre de las carreras de biología marina y ambiental que cursan la materia biología molecular en la Universidad de Bogotá Jorge Tadeo Lozano a diseñar un método diagnóstico para el virus COVID-19 basado en RT PCR y qPCR. El objetivo de este ejercicio académico consistió en incentivar a los estudiantes para que propusieran un método efectivo que permita detectar el virus covid-19 a través de los conocimientos adquiridos durante el curso. Se realizó una revisión de la literatura previa para identificar los métodos existentes para la detección del virus y con ayuda de herramientas bioinformáticas se realizó el análisis de las secuencias de los genomas del COVID-19, el sars humano y el sars de murciélago disponibles en GenBank.
Palabras clave: RT-qPCR,SARS COV 2, pandemia, coronavirus, educación contextualizada.
Abstract: As a pedagogical form of active learning, topics developed in molecular biology class were related to the coronavirus COVID-19 pandemic that is currently advancing in almost the entire planet. Having an efficient method of diagnosing infection during the onset of this acute respiratory infection is important in detecting both symptomatic and asymptomatic patients. For this reason, seventh semester students from marine and environmental biology courses studying molecular biology at the University of Bogotá Jorge Tadeo Lozano were challenged to design a diagnostic method for the COVID-19 virus based on RT PCR and qPCR. The objective of this academic exercise was to encourage students to propose an effective method to detect the COVID-19 virus using the knowledge acquired during the course. A review of the previous literature was carried out to identify the existing methods for the detection of the virus and, with the help of bioinformatic tools, the analysis of the sequences of the genomes of COVID-19, human sars and bat sars available at GenBank was carried out.
Keywords: RT-qPCR, SARS COV 2, pandemic, coronavirus, contextualized education.
Artículos
Integración de herramientas bioinformáticas y métodos en biología molecular para el diseño de un kit diagnóstico del COVID-19: un ejemplo de aprendizaje significativo
Integration of Bioinformatics Tools and Molecular Biology Methods for the design of a COVID-19 Test Kit: An Example of Significant Learning
Universidad de Bogotá Jorge Tadeo Lozano
Recepción: 10 Marzo 2020
Aprobación: 30 Marzo 2020
La biología molecular y la genética en la sociedad del conocimiento del siglo XXI se mezclan cada vez más con disciplinas tradicionalmente separadas pero relacionadas, como la física, la informática y la matemática (Furge, Stevens-Truss, Moore, & Langeland, 2009). El proyecto genoma humano, iniciado en 1990, produjo el desarrollo de una ciencia sin la cual la gran cantidad de datos del genoma no hubiera podido ser analizada: la bioinformática. Esta ciencia –que cada día toma más fuerza–, unida al uso de métodos computacionales, puede resolver problemas biológicos que requieren nuevos enfoques. Un plan de estudio bien estructurado e integral en biología molecular debe incluir esta ciencia. De otra parte, el aprendizaje basado en problemas, orientado a la investigación y el desarrollado en equipos, y dirigido de manera adecuada por profesores, se convierte en una estrategia centrada en el estudiante que apoyan el aprendizaje activo y que son muy efectivas en la educación científica (Prieto, 2006).
La bioinformática se refiere a: “La investigación, desarrollo o aplicación de herramientas y enfoques computacionales para expandir el uso de datos biológicos, médicos, conductuales o de salud, incluidos aquellos para adquirir, almacenar, organizar, archivar, analizar o visualizar datos” (BISTIC, 2000). Teniendo esta definición en mente, y siguiendo nuestro programa durante la semana siete de clases, se desarrolló el tema de métodos en biología molecular, específicamente los métodos de RT-PCR, PCR tiempo real, sondas, hibridaciones de Southern, Northern y Western, así como la utilización de vectores (plásmidos, cósmidos, fásmidos, cromosomas bacterianos y cromosomas de levaduras) para la clonación de genes. Paralelamente, se realizaron aplicaciones bioinformáticas para aprender a descargar secuencias de GenBank y utilizar programas para el corte con enzimas de restricción [Nebcutter V2.0, http://nc2.neb.com/NEBcutter2/] (Vincze, Posfai, & Roberts, 2003) y construir los perfiles electroforéticos. Adicionalmente, los estudiantes aprendieron a realizar alineamientos de secuencias de ADN utilizando ClustalW [https://www.genome.jp/tools-bin/clustalw] (Li, 2003), diseñar primer con Primer3web versión 4.1.0 [http://primer3.ut.ee/] (Untergasser et al., 2012), hacer PCR virtuales con iPCR [https://embnet.vital-it.ch/software/iPCR_form.html] (Lexa, Horak, & Brzobohaty, 2001) y, de manera destacada, a manejar la herramienta BLASTn de GenBank para la búsqueda de secuencias homólogas [https://blast.ncbi.nlm.nih.gov/Blast.cgi] (Altschul, Gish, Miller, Myers, & Lipman, 1990); este último programa además compara secuencias de nucleótidos o proteínas con bases de datos de secuencias y calcula la significación estadística (GenBank, 2020). Teniendo en cuenta lo anterior, se puede afirmar que los estudiantes se relacionaron con las más actuales y modernas herramientas para el aprendizaje de análisis de secuencias proteicas y de ácidos nucleicos.
Históricamente, la educación en ciencia ha requerido desarrollar un enfoque que propicie enseñanza contextualizada o en contexto del mundo real, con lo cual el aprendizaje se convierte en altamente significativo. Así, cuando la nueva información se relaciona con algún aspecto de lo ya existente en la estructura cognitiva del individuo, se produce un proceso que conduce al aprendizaje significativo (Novak, 1988). A partir de este argumento, se introdujo a los estudiantes en el conocimiento de la pandemia producida por el nuevo coronavirus (NCOV 19) descubierto a finales de 2019 en Wuhan, provincia de Hubei, China (Wu et al., 2020). Este coronavirus causa una infección severa en las vías respiratorias. Desde su aparición a principios de diciembre de 2019 hasta el 26 de marzo de 2020, el número de casos ha aumentado exponencialmente hasta 858.785, con un total de 42.151muertes y una recuperación de 178.119 pacientes (Dong, Du, & Gardner, 2020). La pandemia se ha generalizado a 180 países, donde las naciones con mayor número de infectados son China (82.290), Italia (105.792), Estados Unidos (189.035) y España (95.923). Colombia no ha sido ajeno a la pandemia, registrando su primer caso de contagio el 3 de marzo de 2020. Actualmente, el país cuenta con 906 casos reportados (Dong et al., 2020).
Dentro de las políticas que han tomado la mayoría de los países que presentan individuos infectados por covid-19 se encuentran: el aislamiento de los enfermos, la cuarentena de personas que posiblemente han estado expuestas al virus, el confinamiento de todos sus habitantes para prevenir nuevos contagios y la realización de un mayor número de pruebas diagnósticas. Una de estas pruebas se basa en la RT-qPCR, técnica que permite hacer una copia de DNA complementario a partir del RNA viral utilizando una enzima denominada transcriptasa inversa, la cual convierte el RNA de cadena sencilla en DNA de doble cadena. Con esta enzima es posible amplificar diferentes regiones del virus para posibilitar su caracterización. Para ello, se usan primers, o cadenas sencillas cortas de ADN (20 nucleótidos), y sondas de DNA marcadas con fluorocromos para identificar en tiempo real la amplificación de los fragmentos.
La PCR en tiempo real (o cuantitativa) permite medir la cantidad de copias del DNA viral en la muestra. Esta técnica es bastante confiable para identificar los virus. Sin embargo, cuenta con algunas limitaciones. Una de ellas es el tiempo necesario para su ejecución (4-6 h), lo que limita el número de pruebas que se pueden realizar al día. Otra limitante son los reactivos o kits para desarrollar las pruebas, puesto que en este momento se reporta escasez de estos elementos debido a la gran demanda a escala global. Además, la contaminación es un problema limitante, puesto que se requieren laboratorios con suficiente bioseguridad a fin de evitar que se presenten falsos positivos y falsos negativos, como ocurre en algunas ocasiones.
Por otra parte, estas pruebas permiten detectar pacientes asintomáticos, configurando un diagnóstico de gran importancia debido a que estos son transmisores activos de la infección (Costa, 2004; Salazar, Sandoval, & Armendáriz, 2013).
A partir de lo anterior, este trabajo tuvo como objetivo diseñar una prueba diagnóstica con los estudiantes del curso de biología molecular 2020-1 de la Universidad Jorge Tadeo Lozano, quienes trabajaron muy seriamente y propusieron las bases para desarrollar una prueba que podría convertirse en kit para identificar el COVID-19.
Los estudiantes consultaron literatura relacionada con las técnicas moleculares actualmente utilizadas para la identificación de virus de rna en las bases de datos ScienceDirect, Scopus, GenBank y Elsevier. Con base en la información recopilada, se planteó una metodología alternativa para la detección del SARS-COV2. La figura 1 presenta la propuesta metodológica para el desarrollo de este estudio.
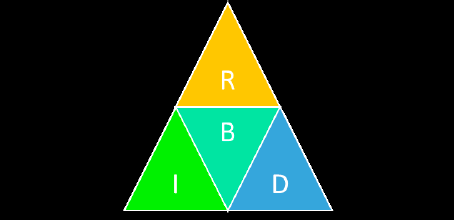
Figura 1
Esquema metodológico utilizado para el desarrollar un método alternativo para detectar el COVID-19
Nota: R) reconocimiento de métodos moleculares y herramientas bioinformáticas para detección y análisis de virus de rna; B) búsqueda de genomas de virus de interés; I) identificación de genes candidatos; D) diseño de primers y sondas.
Fuente: elaboración propia.Se recuperaron de la base de datos de GenBank (NCBI, 2020) los genomas completos de tres cepas de coronavirus correspondientes a Wuhan ncov-19 (NC_045512.2), Human SARS (NC_004718.3) y Bat SARS (DQ412043.1). Las secuencias de cada uno de los 11 genes que componen al virus Wuhan ncov-19 fueron alineadas al genoma de las otras dos especies con ClustalW. Adicionalmente, se seleccionaron los genes que presentaron menor puntaje contra regiones del genoma del Human SARS y Bat SARS como posibles candidatos para la identificación molecular de COVID-19.
Utilizando la secuencia de los genes candidatos identificados previamente mediante ClustalW que presentaron un score inferior a 70 %, se diseñaron primers con el programa Primer3 versión 4.1.0, empleando los parámetros %GC (40 < %GC < 60) y la probabilidad de hair pins (horquillas).
Se diseñaron sondas tipo TagMan a partir de unas regiones de 20 y 24 pb al interior de cada uno de losgenes predichos como posibles marcadores moleculares.Se utilizó el programa MegaX para extraer la secuencia reverse complement de las regiones de cada uno de los genes, las cuales, por complementariedad, se hibridarán a los genes que se quiere identificar. Alas sondas creadas se les incorporará dos fluorocromos (un aceptor y un donador).
Para el diagnóstico del virus COVID-19 se realizará una RT-qPCR, la cual, inicialmente, sintetizará una cadena de CDNA a partir del RNA viral, para posteriormente amplificar y cuantificar en tiempo real la expresión de genes que permitirán su identificación de forma diferencial (Pinilla, 2019).
El SARS-CoV-2 es un betacoronavirus responsable de la pandemia de COVID-19. Este posee un transcriptoma altamente complejo debido a numerosos eventos de recombinación, tanto canónicos como no canónicos. Su genoma está compuesto por 11 genes con longitudes entre 114 y 3.822 pb y comprende cerca de 30.000 pb, distribuido de la siguiente manera: en el extremo 3” una cápside, en el extremo 5” una cola poliA, en su interior dos orfs 1a y 1b, los cuales se traducen en 46 proteínas una vez colonizan la célula, 3 regiones de arn que codifican para proteínas estructurales (“S” proteína espiga “spike protein”, “M” proteína de membrana, “N” proteína de la cápside) y 6 proteínas accesorias (3a, 6, 7a, 7b, 8 y 10), como se muestra en la figura 2 (Kim et al., 2020).
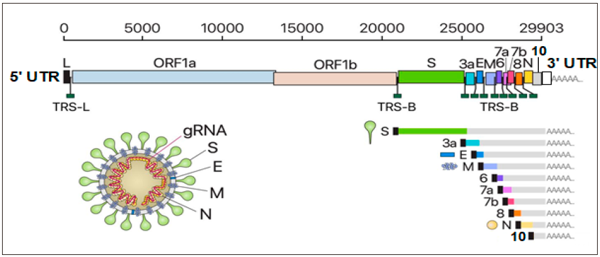
Figura 2
Presentación esquemática de la organización del genoma del SARS-CoV-2 los ARNm subgenómicos canónicos y la estructura del virión
Nota: las cajas grandes representan los orfs, las cajas pequeñas las proteínas accesorias y el cuadro negro indica la secuencia líder.
Fuente: adaptado de Kim et al. (2020).La reacción en cadena de la polimerasa en tiempo real (RT-qPCR) es una metodología sensible que se utiliza ampliamente en diagnóstico e investigación biológica. Su mayor virtud es poder amplificar fragmentos pequeños a partir de mínimas cantidades de DNA del cual se quiere identificar presencia en una muestra. En diagnóstico clínico, la RT-qPCR es utilizada para medir las cargas virales o bacterianas. Por esta razón, los autores consideran que este método es el mejor, más rápido y económico para hacer diagnóstico del coronavirus COVID-19. Sin embargo, se requiere tener alguna experiencia en el manejo de las herramientas bioinformáticas para reconocer los genes o regiones a amplificar, el diseño de primers y el diseño de sondas. La RT-qPCR es utilizada para mejorar la eficiencia diagnóstica como método rápido y temprano para identificar el agente etiológico (en este caso, el coronavirus COVID-19) y de esta manera tomar las medidas de prevención y control del contagio y la enfermedad en fases tempranas con el objetivo de evitar su dispersión (Corman et al., 2020).
El COVID-19 es un virus cuyo material genético es RNA de cadena sencilla. Por ello, se presenta un inconveniente, pues la PCR amplifica únicamente fragmentos de cadena doble, es decir, DNA. Sin embargo, métodos como la RT-PCR (reacción en cadena de la polimerasa con trasncriptasa inversa o reversa) representan una solución para estos casos debido a que permiten convertir una cadena sencilla de RNA en una doble cadena de DNA complementaria (CDNA) con una alta fidelidad (Costa, 2004). En la figura 3 se presenta el proceso mediante el cual, empleando un kit de aislamiento para RNA total, se obtiene el material genético del virus. Posteriormente, utilizando un primer de TTTTs, se origina la primera cadena de CDNA y luego, por PCR, se obtienen cadenas dobles del CDNA del virus (figura 3).
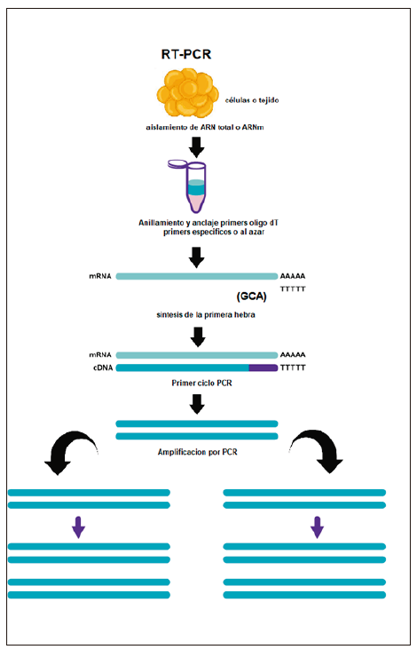
Figura 3
Proceso de RT-PCR por el cual se obtiene una cadena doble de DNA complementario
Fuente: adaptada de https://www.sigmaaldrich.com/life-science/molecular-biology/pcr/rt-pcr.html.
Al tener la cadena de CADN se prosigue con la amplificación de uno o varios fragmentos de esta molécula. El COVID-19 tiene un RNA de 29,903 pb (figura 1). La qPCR es un método que amplifica regiones o fragmentos de ADN y los detecta de manera paralela dentro del mismo microtubo utilizado. Por ello, se utilizan sondas, que son fragmentos marcados de ADN de cadena sencilla complementarios a las zonas que se van a amplificar, de manera que al unirse a los fragmentos amplificados permite identificarlos y cuantificarlos. Los autores del presente trabajo seleccionaron sondas de hidrólisis (Taqman) que corresponden a fragmentos de ADN de cadena sencilla marcados en el extremo 3’ con un fluorocromo donador que reporta la fluorescencia, emitiendo luz al ser chocado por la ADN polimerasa, y un aceptor en el extremo 5’, llamado quencher, el cual atrapa la fluorescencia que libera el donador. Las moléculas donadora y aceptora deben estar cercanas, de manera que cuando la sonda está intacta, la fluorescencia emitida por el donador no es observable, al ser absorbida por el aceptor (figura 4A). Durante la amplificación, la Taq polimerasa de ADN rompe o libera el extremo libre 5’ de la sonda que contiene al donador al iniciar la síntesis, permitiéndole emitir la florescencia (figura 4B), que es captada por el lector (figura 4C) (Costa, 2004).
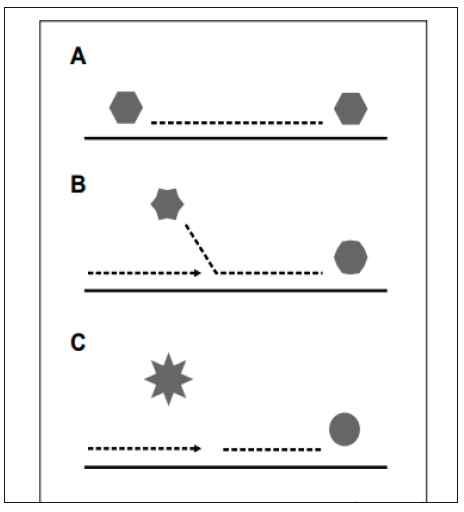
Figura 4
Proceso e activación de la fluorescencia en la qPCR
Nota: A) la sonda se hibrida a la zona complementaria del fragmento que se va a amplificar, el extremo donador está cerca al quencher, no hay florescencia; B) la polimerasa que ha iniciado la síntesis de DNA choca contra el donador y lo libera, produciéndose florescencia; C) la florescencia es captada y trasmitida por el lector.
Fuente: tomada de Costa (2004).Las secuencias de los genomas de Wuhan nCOV-19, Human SARS y Bat SARS descargados de GenBank presentaron longitudes de 29.903, 29.751 y 29.749 pb, respectivamente. El virus Wuhan- nCOVID-19, estructuralmente está compuesto por 11 genes distribuidos como se presenta en la tabla 1, con longitudes entre 114 y 3.822 pb. A diferencia del SARS humano, el SARS aislado en murciélagos es 0,5 % más largo y su composición está bien definida a nivel de UTRs, CDs y proteínas.
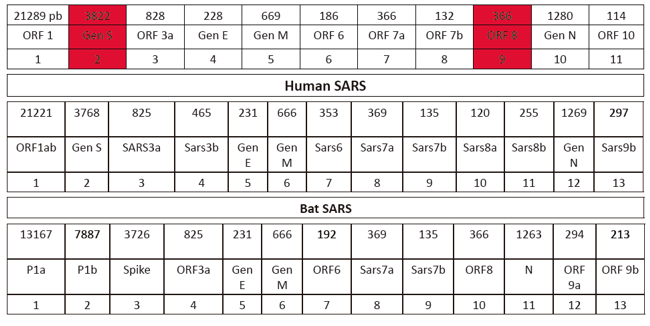
Tabla 1
Composición estructural de los genes del virus Wuhan n-COVID-19, Human SARS y Bat SARS utilizados en este estudio
Las secuencias de los once genes presentados por el Wuhan COVID-19 coronavirus (NC_045512.2) fueron alineados con los genomas de las cepas de Human-SARS y Bat-SARS mediante ClustalW. Los puntajes obtenidos por el alineamiento de cada una de las secuencias con cada uno de los 11 genes coincidió con que los genes 2 y 9 presentes en COVID-19 podrían ser utilizados para su identificación por RT-qPCR, considerando que los dos alineamientos presentaron puntajes menores a 70 % contra regiones de los dos coronavirus (figuras 5 y 6). Los alineamientos de estos dos genes al genoma del virus Human-SARS se pueden apreciar en las figuras 7 y 8.
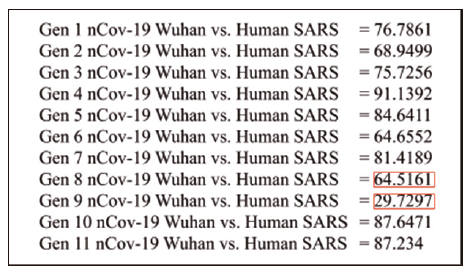
Figura 5
Puntaje de alineamiento (%) de los 11 genes presentes en el Wuhan nCOV-19 vs. el genoma de Human SARS
Nota: los puntajes en el recuadro rojo corresponden a los genes candidatos para el diagnóstico.
Fuente: elaboración propia.
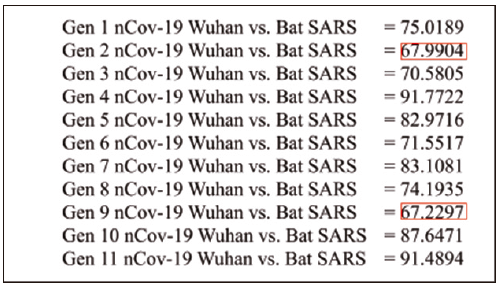
Figura 6
Puntaje de alineamiento (%) de los 11 genes presentes en el Wuhan nCOV-19 vs. el genoma de Bat SARS
Nota: los puntajes en el recuadro rojo corresponden a los genes candidatos para el diagnóstico.
Fuente: elaboración propia.

Figura 7
Human sars vs. gen 9 (orf 8) del nCoV-19 (Wuhan)
Fuente: elaboración propia mediante ClustalW (Aligned score =29,72).

Figura 8
Alineamiento genoma Human SARS (NC_004718.3), vs. gen 2 (ORF 8) del virus nCoV-19 (Wuhan, NC_045512.2).
Fuente: elaboración propia mediante ClustalW (Aligned score = 67,99).







Utilizando el programa Primer3 versión 4.1.0 y la secuencia completa de los dos genes candidatos (genes 2 y 9) como posibles marcadores moleculares para la identificación de covid-19, se diseñaron dos pares de primers con una longitud de 20 pb cada uno, los cuales amplifican en su totalidad cada uno de los genes. Sus características se presentan en la tabla 2.

Tabla 2
Características de los primers diseñados para la identificación de COVID-19
A pesar de que el gen número 8 también produjo una puntuación baja, se descartó su uso debido a que este correspondía a una secuencia muy corta dentro del genoma del Wuhan nCOV-19 (figura 1) y por tanto no cumple con las características para el diseño de primers.
Para el diseño de las sondas se seleccionaron 2 regiones dentro de los genes 2 y 9, de 20 y 24 pb, respectivamente. Con ayuda del programa MegaX, se obtuvieron las cadenas complementarias a esas regiones utilizando la opción reverse complement. Las regiones seleccionadas se hibridarán por complementariedad a los genes que se requiere identificar. Además, a estas sondas se les incorporará dos tipos de fluorocromos (un aceptor y un donador). Las sondas diseñadas se presentan a continuación:
Sonda COVID-19- Gen2 (Size = 20pb)

Sonda COVID-19- Gen9 (Size = 24pb)

Una vez tenemos los primers y las sondas diseñadas para amplificar un fragmento de los genes S y ORF8, es posible afirmar que contamos con los componentes necesarios para realizar la identificación del COVID-19; los demás reactivos, como enzimas, tampones y cofactores, se consiguen fácilmente en el mercado.
Se propuso un diseño para el desarrollo de la reacción de RT-qPCR y de esta forma identificar el virus covid-19 responsable de la pandemia que actualmente se desarrolla en todo el planeta. Para ello, se utilizaron métodos moleculares (RT-qPCR) y programas bioinformáticos que permitieron analizar las secuencias del virus y a partir de ellas diseñar los primers y sondas para su amplificación y cuantificación. Por medio de este ejercicio, los estudiantes lograron comprender las variables de las dos reacciones: RT-PCR y la PCR Tiempo Real.
Aunque, en general, se utilizaron de forma adecuada las herramientas bioinformáticas para encontrar los genes, diseñar los primers y las sondas, se presentó un error conceptual, puesto que al inicio se realizaron las comparaciones entre genes y genomas. No obstante, se hizo necesario comparar gen con gen para detectar la identidad de cada uno de ellos y así detectar zonas de homología y zonas variables, con el fin de diseñar los primers de tal manera que se aseguraran de amplificar fragmentos únicos del COVID-19. Finalmente, esto se consiguió gracias a que los estudiantes aprendieron del error, lo corrigieron y lograron identificar los genes más variables.
ClustalW es una herramienta sencilla que permite explicar conceptos básicos de biología molecular y hacer comparaciones entre secuencias equidistantes y de una longitud similar (Thompson, Plewniak, & Poch, 1999). Al realizar comparaciones entre secuencias de diferente longitud, y con base en la plataforma que se utilice, el resultado de los alineamientos difiere, ya que cada plataforma maneja sus propios parámetros por defecto. Por ende, cuando se realizan alineamientos múltiples se asignan diferentes puntuaciones a los gaps e inserciones para optimizarlos (Gaskell, 2000).
La realización de este tipo de ejercicios académicos por parte de los estudiantes de biología molecular permite afianzar los conocimientos adquiridos, incentivar la creatividad y gusto por las ciencias ómicas y despertar su curiosidad por comprender nuevas herramientas como la bioinformática, lo cual nos permite adentrarnos y comprender nuevos conceptos y procesos relacionados con la vida, contribuyendo a descubrir los misterios ocultos de la naturaleza.
Se logró el objetivo planteado para esta investigación al analizar las secuencias de los virus y diseñar primers y sondas, los cuales son los insumos más importantes para realizar el diagnóstico de COVID-19.
El trabajo presentado consistió en la aplicación de la metodología de aprendizaje basado en problemas (ABP) a manera de un ejercicio de aprendizaje en contexto, el cual fue llevado al plano actual de la pandemia de COVID-19 con un enfoque interdisciplinar en el que se debían aplicar conocimientos teóricos de métodos en biología molecular y emplear diferentes herramientas bioinformáticas.
ClustalW y primers 3 son dos herramientas bioinformáticas fáciles de usar que resultan muy valiosas, ya que permiten a los estudiantes afianzar los conocimientos adquiridos en clase y desarrollar su capacidad de análisis.
Ejercicios académicos como estos ayudan a los estudiantes a comprender la importancia de la biología molecular, la bioinformática y las ciencias ómicas para el desarrollo de estrategias que permitan detectar virus emergentes y contribuir al desarrollo de la ciencia.
Figura 1
Esquema metodológico utilizado para el desarrollar un método alternativo para detectar el COVID-19
Nota: R) reconocimiento de métodos moleculares y herramientas bioinformáticas para detección y análisis de virus de rna; B) búsqueda de genomas de virus de interés; I) identificación de genes candidatos; D) diseño de primers y sondas.
Fuente: elaboración propia.Figura 2
Presentación esquemática de la organización del genoma del SARS-CoV-2 los ARNm subgenómicos canónicos y la estructura del virión
Nota: las cajas grandes representan los orfs, las cajas pequeñas las proteínas accesorias y el cuadro negro indica la secuencia líder.
Fuente: adaptado de Kim et al. (2020).Figura 3
Proceso de RT-PCR por el cual se obtiene una cadena doble de DNA complementario
Fuente: adaptada de https://www.sigmaaldrich.com/life-science/molecular-biology/pcr/rt-pcr.html.
Figura 4
Proceso e activación de la fluorescencia en la qPCR
Nota: A) la sonda se hibrida a la zona complementaria del fragmento que se va a amplificar, el extremo donador está cerca al quencher, no hay florescencia; B) la polimerasa que ha iniciado la síntesis de DNA choca contra el donador y lo libera, produciéndose florescencia; C) la florescencia es captada y trasmitida por el lector.
Fuente: tomada de Costa (2004).Tabla 1
Composición estructural de los genes del virus Wuhan n-COVID-19, Human SARS y Bat SARS utilizados en este estudio
Figura 5
Puntaje de alineamiento (%) de los 11 genes presentes en el Wuhan nCOV-19 vs. el genoma de Human SARS
Nota: los puntajes en el recuadro rojo corresponden a los genes candidatos para el diagnóstico.
Fuente: elaboración propia.Figura 6
Puntaje de alineamiento (%) de los 11 genes presentes en el Wuhan nCOV-19 vs. el genoma de Bat SARS
Nota: los puntajes en el recuadro rojo corresponden a los genes candidatos para el diagnóstico.
Fuente: elaboración propia.Figura 7
Human sars vs. gen 9 (orf 8) del nCoV-19 (Wuhan)
Fuente: elaboración propia mediante ClustalW (Aligned score =29,72).
Figura 8
Alineamiento genoma Human SARS (NC_004718.3), vs. gen 2 (ORF 8) del virus nCoV-19 (Wuhan, NC_045512.2).
Fuente: elaboración propia mediante ClustalW (Aligned score = 67,99).
Tabla 2
Características de los primers diseñados para la identificación de COVID-19


