

Artículos científicos
LA QUÍMICA COMPUTACIONALCOMO HERRAMIENTA PARA ENTENDERPROCESOS QUÍMICOS Y BIOQUÍMICOS A NIVEL MOLECULAR
COMPUTATIONAL CHEMISTRY AS A TOOL TO UNDERSTAND CHEMICAL AND BIOCHEMICAL PROCESSES AT A MOLECULAR LEVEL
infoANALÍTICA
Pontificia Universidad Católica del Ecuador, Ecuador
ISSN: 2602-8344
ISSN-e: 2477-8788
Periodicidad: Semestral
vol. 8, núm. Esp.2, 2020
Recepción: 09 Julio 2020
Aprobación: 30 Octubre 2020
Resumen: Con el avance de la tecnología, la química computacional se ha convertido en pieza clave de las investigaciones científicas. Hoy en día, la química computacional ha logrado obtener métodos y algoritmo capaces de reducir errores en la predicción de propiedades y simulación de eventos químicos llegando a ser comparables con resultados experimentales. Ser capaces de entender diferentes procesos químicos desde un punto de vista molecular, lo que muchas veces es imposible conseguir mediante la experimentación, hace de la química computacional una herramienta poderosa. Así, procesos como el descubrimiento de nuevos medicamentos, han dejado de depender del factor del azar, para transformarse en procesos más eficientes y racionales llegando a reducir el tiempo en un 50 %. En esta revisión, examinaremos algunos ejemplos de cómo la química computacional ha ayudado a tener un mejor entendimiento de procesos químicos como la sustitución nucleofílica de Vicarius, la interacción de nanopartículas de plata con moléculas orgánicas y el mecanismo de reacción de la biodegradación de la lignina en presencia de la lignina peroxidasa. También se presenta investigaciones sobre el posible mecanismo de acción de fármacos como el paracetamol o de los péptidos antimicrobianos extraídos del exudado de ranas.
Palabras clave: DFT, mecanismos de reacción, péptidos antimicrobianos, química computacional, reactividad.
Abstract: As technology advances, computational chemistry has become a key part of scientific research. Nowadays, computational chemistry has been able to obtain methods and algorithms capable of reducing errors in the prediction of properties and simulation of chemical events being comparable with results obtained experimentally. Being able to understand different chemical processes from a molecular point of view, which sometimes is impossible to achieve through experimentation, makes computational chemistry a powerful tool. In this sense, processes such as drug discovery have ceased to depend on serendipity to become a more efficient and rational process; therefore, reducing the time to put a drug in clinical trials by half. In this review, some examples of how computational chemistry has helped to get a better understanding of chemical processes are examined. Studies getting new insights into the Vicarious nucleophilic substitution, interaction of silver nanoparticles with organic molecules, and the reaction mechanism of lignin biodegradation in the presence of lignin peroxidase are reviewed. Also, investigations on the possible mechanism of action of drugs such as acetaminophen or antimicrobial peptides extracted from frog exudate are presented.
Keywords: antimicrobial peptides, computational chemistry, DFT, reaction mechanism, reactivity.
RESUMEN
Con el avance de la tecnología, la química computacional se ha convertido en pieza clave de las investigaciones científicas. Hoy en día, la química computacional ha logrado obtener métodos y algoritmo capaces de reducir errores en la predicción de propiedades y simulación de eventos químicos llegando a ser comparables con resultados experimentales. Ser capaces de entender diferentes procesos químicos desde un punto de vista molecular, lo que muchas veces es imposible conseguir mediante la experimentación, hace de la química computacional una herramienta poderosa. Así, procesos como el descubrimiento de nuevos medicamentos, han dejado de depender del factor del azar, para transformarse en procesos más eficientes y racionales llegando a reducir el tiempo en un 50 %. En esta revisión, examinaremos algunos ejemplos de cómo la química computacional ha ayudado a tener un mejor entendimiento de procesos químicos como la sustitución nucleofílica de Vicarius, la interacción de nanopartículas de plata con moléculas orgánicas y el mecanismo de reacción de la biodegradación de la lignina en presencia de la lignina peroxidasa. También se presenta investigaciones sobre el posible mecanismo de acción de fármacos como el paracetamol o de los péptidos antimicrobianos extraídos del exudado de ranas.
ABSTRACT
As technology advances, computational chemistry has become a key part of scientific research. Nowadays, computational chemistry has been able to obtain methods and algorithms capable of reducing errors in the prediction of properties and simulation of chemical events being comparable with results obtained experimentally. Being able to understand different chemical processes from a molecular point of view, which sometimes is impossible to achieve through experimentation, makes computational chemistry a powerful tool. In this sense, processes such as drug discovery have ceased to depend on serendipity to become a more efficient and rational process; therefore, reducing the time to put a drug in clinical trials by half. In this review, some examples of how computational chemistry has helped to get a better understanding of chemical processes are examined. Studies getting new insights into the Vicarious nucleophilic substitution, interaction of silver nanoparticles with organic molecules, and the reaction mechanism of lignin biodegradation in the presence of lignin peroxidase are reviewed. Also, investigations on the possible mechanism of action of drugs such as acetaminophen or antimicrobial peptides extracted from frog exudate are presented.
INTRODUCCIÓN
Hoy en día, las computadoras se han convertido en parte fundamental de las ciencias básicas como la física, química y matemáticas. La química computacional es una rama de la química que usa modelos matemáticos implementados en computadoras para facilitar y entender con más profundidad diferentes procesos químicos (Foresman, 2001). La química computacional puede ser aplicada en diversas áreas como como la bioquímica, ciencia de materiales, nanotecnología y biología molecular. Durante los últimos 30 años, varias herramientas han sido desarrolladas permitiendo, desde, analizar el gran volumen y complejidad de datos generados hasta modelar procesos químicos (Fourches, Muratov, & Tropsha, 2010; Hassan, Brown, Varma-O’Brien, & Rogers, 2006).
El modelamiento molecular trata de imitar el comportamiento de las moléculas estudiando, así, desde pequeños sistemas orgánicos en fase gaseosa hasta el estudio de complejas interacciones químicas en sistemas biológicos (Holtje, Folkers, & Luzar, 1998; Karlström et al., 2003).
Dependiendo del problema que se quiera estudiar, la química computacional ofrece una gran variedad de algoritmos teóricos y métodos para abarcarlos como son los mecánico cuánticos, estadísticos, ab initio, dinámica molecular, funcionales semiempíricos y de densidad, entre otros (Lewars & Lewars, 2016a). Con estos métodos, se pueden calcular geometrías moleculares, tasas y equilibrios, reactividad, espectros, mecanismos de reacción, interacciones moleculares, modelos relación cuantitativa estructura-actividad (QSAR por sus siglas en inglés) y otras propiedades físicas de interés (Fourches et al., 2010; Hassan et al., 2006; Lewars & Lewars, 2016b, 2016c, 2016a).
Para el estudio de moléculas pequeñas, se utilizan modelos cuanto mecánicos sofisticados y costosos computacionalmente (Lewars & Lewars, 2016b). La mecánica cuántica es la base de la física moderna y parte esencial de la fisicoquímica. Tuvo sus inicios en la radiación del cuerpo negro y el efecto fotoeléctrico, llegando al átomo de Bohr, y finalizando con la base de la mecánica cuántica que es la ecuación de Schrödinger (ecuación 1). Esta ecuación permite predecir el comportamiento de un sistema dinámico, aunque sólo se ha logrado resolver para sistemas pequeños como el átomo de hidrógeno (Bell, Dines, Chowdhry, & Withnall, 2007).
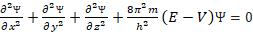 (1)
(1)Sistemas un poco más grandes son estudiados con modelos que usan cierto tipo de aproximaciones para no hacerlos tan demandantes computacionalmente como es la teoría del funcional de la densidad (ecuación 2) (Lewars & Lewars, 2016a). Sistemas biológicos mucho más grandes y que tomarían tiempos inmanejables usando los métodos anteriormente mencionados, son estudiados con métodos más simples como la mecánica clásica (Karlström et al., 2003).
 (2)
(2)La química computacional es ampliamente utilizada en la industria farmacéutica para explorar las interacciones de posibles fármacos con biomoléculas, por ejemplo mediante el acoplamiento de un candidato de fármaco con en el sitio activo de su objetivo biológico (Lewars & Lewars, 2016a, 2016c). También, esta rama de la química tiene gran aplicación en la investigación de las propiedades de sólidos y polímeros siendo fundamental en la fabricación de nuevos materiales (Lewars & Lewars, 2016a). Se debe enfatizar que ninguno de estos cálculos puede reemplazar a los trabajos experimentales que siguen teniendo la palabra final de lo que en verdad sucede en la naturaleza. Sin embargo, la química computacional es un complemento para entender más a profundidad eventos que son imposibles de visualizar experimentalmente (Lewars & Lewars, 2016a).
En esta revisión, se presentarán varios de los trabajos realizados por el grupo de química computacional de la Pontificia Universidad Católica del Ecuador, donde se utilizaron varias de las metodologías computacionales disponibles para estudiar propiedades como reactividad, mecanismos de reacción, mecanismos de acción de fármacos y péptidos. Los resultados obtenidos permitieron entender de mejor manera procesos que envuelven tanto moléculas pequeñas, así como sistemas enzimáticos.
RESULTADOS
Reactividad
La reactividad es un parámetro muy usado en química para determinar qué tan fácil es para una molécula experimentar una reacción química. Saber la reactividad de cierto tipo de moléculas en reacciones de interés como la sustitución nucleofílica de Vicarius (SNV) es de gran importancia para entender este tipo de proceso y poder obtener productos deseados de una forma más eficiente. La SNV es una herramienta útil para introducir una variedad de sustituyentes en compuestos aromáticos y heteroaromáticos (Ma̧kosza & Wojciechowski, 2004). Varios estudios experimentales se han llevado a cabo para sintetizar nuevos nitroarenos sustituidos y para determinar las constantes de velocidad cinética de estas reacciones (Mąkosza, 2010; Ma̧kosza & Wojciechowski, 2004; Patz, Mayr, Maruta, & Fukuzumi, 1995). Esta reacción es ampliamente utilizada ya que permite introducir diferentes grupos funcionales conteniendo átomos de carbono en anillos nitroaromáticos (Vollhardt & Schore, 2014). En general, la reacción se realiza mediante la adición al nitroareno de un carbanión con un grupo saliente X unido a su centro, seguido de una eliminación β del HX como se muestra en el Esquema 1 (Ma̧kosza & Wojciechowski, 2004). Varios estudios se han realizado para entender y aclarar varias de las características esenciales de la reacción por su amplia aplicabilidad en síntesis orgánica (Mąkosza, 2010; Ma̧kosza, Lemek, Kwast, & Terrier, 2002; Ma̧kosza & Wojciechowski, 2004; Patz et al., 1995; Vollhardt & Schore, 2014), aunque su mecanismo de reacción aún no está del todo elucidado debido a la complejidad experimental de las investigaciones cinéticas (Mąkosza, 2010). La naturaleza del solvente, el tipo de nucleófilo, la demanda estérica del cabanión, la concentración y naturaleza de la base tienen una influencia considerable tanto en la energía de activación como en la relación isomérica de los productos que es importante estudiarla para conocer la influencia de cada uno (Patz et al., 1995).
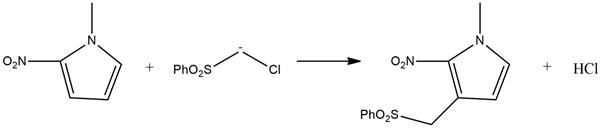
En estudios teóricos, la Teoría del Funcional de la Densidad (DFT) ha sido exitosa al proporcionar un conjunto de índices de reactividad global y local para el análisis de selectividad química y reactividad en numerosos sistemas (Winiarski & Makosza, 1987). Estos índices son obtenidos en términos de parámetros de reactividad global como el potencial químico electrónico, μ; dureza química, h y suavidad global, S (Schindele, Houk, & Mayr, 2002). Adicionalmente, descriptores de selectividad como la función Fukui, ¦(r) y suavidad local, s(r) (Schindele et al., 2002; Winiarski & Makosza, 1987). Uno de los parámetros de reactividad más usados en DFT es la electrofilia global, w, definida por Parr. (Parr, 1980). Este descriptor ha sido utilizado con éxito para describir la reactividad en diferentes sistemas orgánicos como reacciones de Diels-Alder, las cicloadiciones 1,3-dipolares y las reacciones de Friedel-Crafts (Parr, Szentpály, & Liu, 1999; Parr & Yang, 1984; Pérez, Domingo, Aurell, & Contreras, 2003).
En este primer proyecto, se analizaron los patrones de reactividad de una serie de arenos deficientes en electrones, utilizando el índice de electrofilia global (Parr, 1980). y un nivel de teoría B3LYP/6-311G(d,p) implementado en el software Gaussian 09 (Frisch et. al, 2009). Los valores obtenidos computacionalmente fueron comparados con valores experimentales obtenidos en la literatura (Mąkosza, 2010; Ma̧kosza et al., 2002; Ma̧kosza & Wojciechowski, 2004).
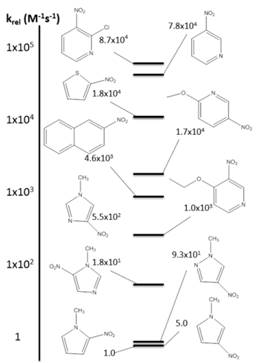
La velocidad relativa de segundo orden obtenida experimentalmente fue comparada con los valores de electrofilia global obtenidos computacionalmente. Ya que la relación entre los dos es logarítmica, el logaritmo de la constante de velocidad fue obtenido (Tabla 1). Los compuestos estudiados por Meneses et.al (2017), fueron divididos en dos grupos dependiendo del número de átomos que conforma el anillo. Los valores obtenidos en la Tabla 1 fueron graficados en la Figura 1, con la obtención de una buena correlación entre ambos parámetros.
| Compuesto | krel (M-1s-1) | Log krel | w(eV) |
| 3-nitropiridina | 7,8x104 | 4,89 | 0,935 |
| 2-cloro-3-nitropiridina | 8,7x104 | 4,94 | 0,921 |
| 4-etoxi-3-nitropiridina | 1,0x103 | 3,00 | 0,550 |
| 2-metoxi-5-nitropiridina | 1,7x104 | 4,23 | 0,758 |
| 1-nitronaftaleno | 4,6x103 | 3,66 | 0,693 |
| N-metil-2-nitropirrol | 1,0 | 0,00 | 0,626 |
| N-metil-3-nitropirrol | 5,0 | 0,70 | 0,502 |
| 1-metil-5-nitroimidazol | 1,8x101 | 1,26 | 0,751 |
| 1-metil-4-nitroimidazol | 5,5x102 | 2,74 | 0,609 |
| 1-metil-4-nitropirazol | 9,3x101 | 1,97 | 0,722 |
| 2-nitrotiofeno | 1,8x104 | 4,26 | 0,918 |
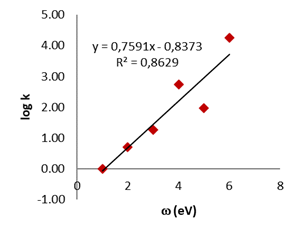
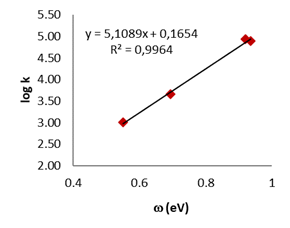
La Figura 1 muestra una línea recta con una tendencia positiva y un coeficiente de correlación de 0,99 para los compuestos que tienen anillos de seis miembros y de 0,86 para los de cinco miembros, indicando una fuerte relación entre ambos parámetros. Esto demuestra que el método DFT usado es óptimo para el estudio de la reactividad en este tipo de sistemas. El descriptor de electrofilia global obtenido computacionalmente puede ser usado para estimar la constante de velocidad de los nitroarenos mediante su relación logarítmica.
La electrofilia global es un excelente descriptor de reactividad, ya que explica adecuadamente los efectos electrónicos de los sustituyentes en las moléculas estudiadas, permitiendo clasificar adecuadamente los nitroarenos en su escalas absolutas de reactividad (Figura 2) (Meneses, Morocho, Castellanos, & Cuesta, 2017).
Mecanismos de reacción
La lignina peroxidasa (LiP) es una enzima encargada de degradar hidrocarburos aromáticos en la naturaleza (Kudo, Harada, Kubota, Sasaki, & Kaneta, 2017). Debido a su eficiencia, este mecanismo ha sido usado también para aplicaciones industriales entre las que se encuentran una gran variedad de campos, incluidos la industria del papel, cosméticos, alimentos, textiles, industrias de combustibles, químicos y productos agrícolas (MacDonald, Goacher, Abou-Zaid, & Master, 2016; Rai, Yadav, & Singh Yadav, 2016). En presencia de H2O2, la LiP puede descomponer fácilmente la lignina y sus análogos sin la necesidad de condiciones agresivas como ácidos, altas temperaturas o presiones. Esto, reduce el aporte de energía, el impacto ambiental y proporciona un alternativa más específica y efectiva (Egwim, Kabiru, & Tola, 2015; Harada, Sasaki, & Kaneta, 2016; Kudo et al., 2017). Estudios han sugerido que estas enzimas pueden también degradar algunos contaminantes orgánicos, como el indano, pentaclorofenol, bifenilos policlorados, pireno, y cianuro (Shah, Grover, Barr, & Aust, 1992).
Varias investigaciones se han llevado a cabo en un intento de obtener una mejor comprensión del complejo proceso de la biodegradación de la lignina por acción de la LiP. Entre esos, se ha caracterizado la enzima en términos de su estructura y propiedades biofísicas y químicas (Ten Have, Franssen, & Field, 2015). Otros estudios se han dirigido hacia la dilucidación de los mecanismos químicos exactos implicados en el rompimiento del enlace de la cadena lateral de la lignina y las reacciones de apertura del anillo aromático (Dashtban, Schraft, Syed, & Qin, 2010).
En el mecanismo de reacción de la degradación de la lignina, esta enzima cataliza el rompimiento del enlace carbono-carbono, degradando así este polímero. Si se parte de otro compuesto orgánico como el 1-(3’,4’-dimetoxifenil)propeno (DMPP), este mecanismo produce veratraldehido (VAD), que es un compuesto ampliamente usado en la industria como saborizante y como punto de partida en la síntesis de fármacos como el Prazosin o la Dimeditiapramine (Cuesta, et. al., 2018).
Uno de los pocos, pero más completos mecanismos de reacción propuestos para este proceso, muestra que durante la transformación ocurren solo dos pasos donde se incluye la LiP, mientras que el resto de los pasos son no enzimáticos. En el mecanismo, un radical libre catiónico del alcohol veratrílico se forma como un intermedio (Khindaria, Yamazaki, & Aust, 1995). Además de la reacción de oxidación de un electrón catalizada por la LiP, este mecanismo pasa por varias reacciones no enzimáticas, incluida la adición de agua y la formación de radicales. Los radicales son especies reactivas que permiten reacciones secundarias, como la adición de un radical superóxido, el acoplamiento o formación de radicales peroxilo (Ten Have et al., 2015). De este modo, se propuso estudiar los diferentes pasos involucrados en el mecanismo de reacción de la LiP (Figura 3) usando DFT para caracterizar los reactivos, intermedios, estados de transición y productos propuesto por Ten Have et al. (2015) Adicionalmente, se realizó una caracterización cinética del mecanismo usando descriptores como la fuerza de reacción y la constante de la fuerza de reacción. Debido a la complejidad del modelo biológico, los pasos donde la LiP participa como catalizador, no fueron tomados en cuenta en este estudio (Cuesta et. al., 2018).
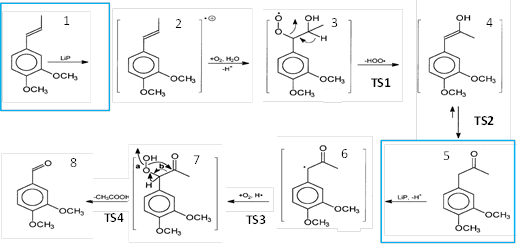
Todos los cálculos teóricos para el estudio del mecanismo de reacción se realizaron utilizando el funcional global hibrido M06-2X y un conjunto de base 6‑31++G(d, p) (Zhao & Truhlar, 2008). El mecanismo de reacción estudiado para la formación de VAD a partir de DMPP comprende siete pasos, de los cuales cinco pasos no incluyen la LiP como catalizador. Estos cinco pasos incluyen la formación de especies radicales, transformación de enlaces, adición de agua y oxígeno, reordenamiento de átomos y una desacetilación. Los resultados de Cuesta et. al., (2018). mostraron que el mecanismo general estudiado que va desde la estructura 3 a la estructura 8 (Figura 4) es un proceso endotérmico, con energía de activación mixta que depende de los cuatro estados de transición encontrados (Tabla 2). Ya que la LiP interviene en la mitad de la reacción, se obtuvieron dos pasos determinantes, uno para los procesos que ocurren antes (paso 2) y uno para los procesos que ocurren después (paso 4) de la intervención de la LiP.
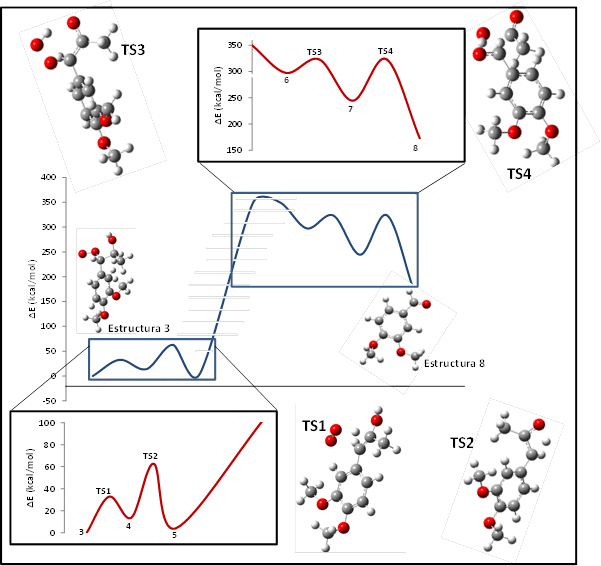
| Paso | TS 1 | TS 2 | TS 3 | TS 4 |
| EA (kcal/mol) | 32.80 | 49.03 | 25.71 | 79.52 |
| HA (kcal/mol) | 32.80 | 49.03 | 25.12 | 79.52 |
| GA (kcal/mol) | 32.92 | 49.36 | 38.37 | 81.20 |
| SA (cal/mol.K) | -0.42 | -1.10 | -44.45 | -5.63 |
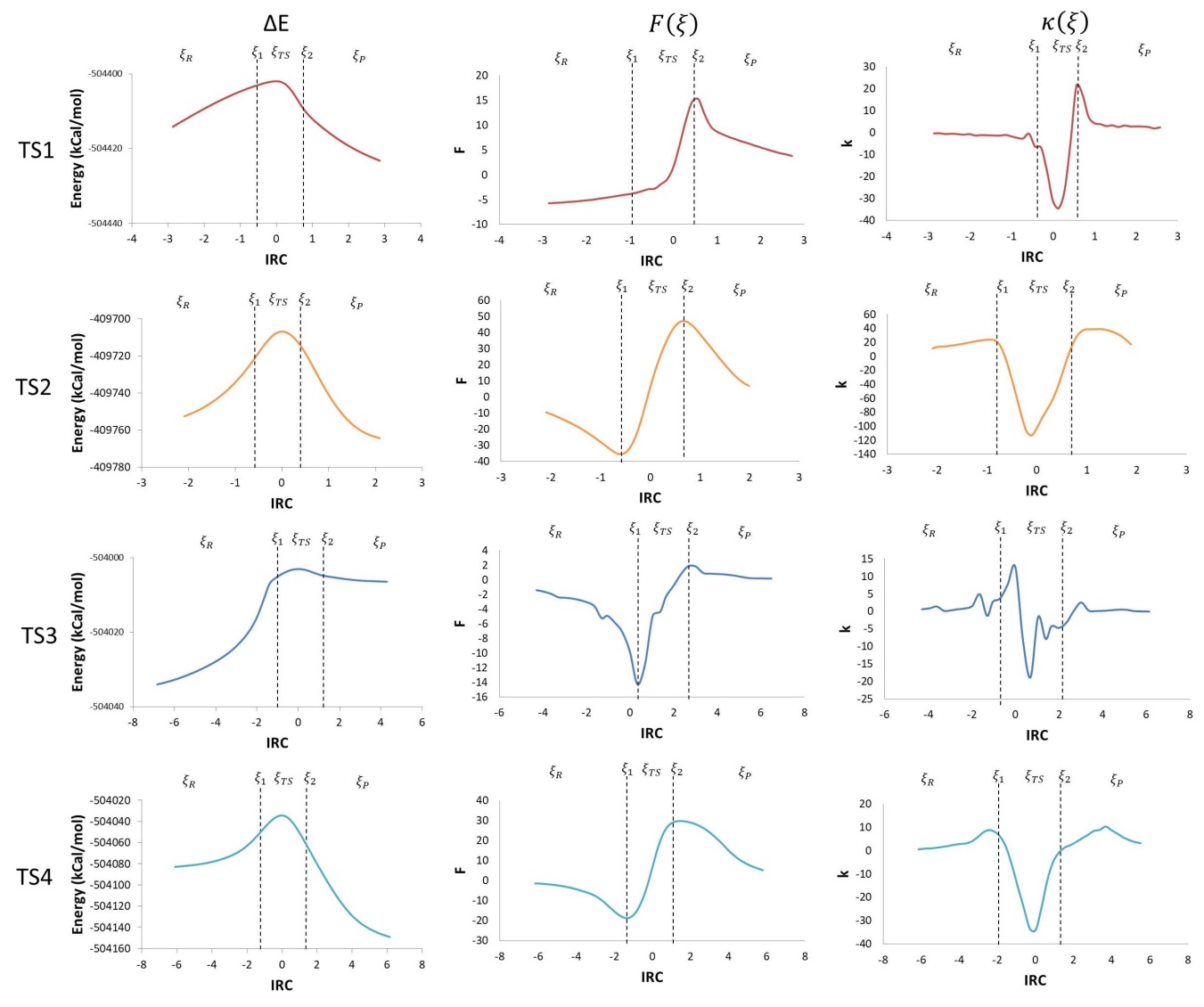
En la Figura 5 se muestran los descriptores de la fuerza de reacción y la constante de fuerza para los pasos determinantes. En ésta, se puede observar cómo para la fuerza de reacción, la región del estado de transición va desde un mínimo global a un máximo, siendo cero en el estado de transición. En contraste, para la constante de fuerza, los valores durante la región del estado de transición son negativos, llegando a su punto mínimo en el estado de transición.
Este estudio preliminar de Cuesta et. al. (2018), aunque consiste en modelos moleculares muy simples y que no involucra efectos del ambiente proteico o de la solvatación explícita de agua en las diferentes etapas de la reacción, es útil para entender a un nivel molecular los pasos donde la LiP no interviene directamente. Estudios se siguen haciendo para crear un modelo molecular completo sustrato-enzima, donde todos los efectos se puedan tomar en cuenta utilizando métodos de dinámica molecular y QM/MM.
Mecanismo de acción
El paracetamol es uno de los medicamentos más usados a nivel mundial. Actualmente, se considera el principal analgésico y antipirético del mercado (Jozwiak-Bebenista & Nowak, 2014). A lo largo de los años, el paracetamol ha estado bajo constante investigación sin poder aclarar o verificar su mecanismo de acción en el cuerpo, lo que ha creado un debate sobre si este medicamento debe ser considerado parte del grupo de medicamentos antiinflamatorios no esteroideos (AINE) (Ottani, Leone, Sandrini, Ferrari, & Bertolini, 2006). Debido a esto, varias investigaciones se han centrado en su relación con las ciclooxigenasas (COX) (Graham, Davies, Day, Mohamudally, & Scott, 2013; Graham & Scott, 2005), lo que ha llevado a sugerir la existencia de una variante adicional, denominada COX-3 (Botting & Ayoub, 2005; Kis, Snipes, Bari, & Busija, 2004; Kis, Snipes, & Busija, 2005; Snipes, Kis, Shelness, Hewett, & Busija, 2005). Sin embargo, las conclusiones de estas investigaciones no han sido decisivas, entregando resultados en los que se evidencia que esta enzima, aunque puede tener actividad COX en caninos y, por lo tanto, interactuar con el Paracetamol, en roedores y humanos, su codificación genera aminoácidos completamente diferentes en comparación a las COX-1 y COX-2, sin mostrar actividad (Kis et al., 2005). Otras hipótesis sugieren que el medicamento actúa en la mediación de los receptores cannabinoides, específicamente el receptor potencial transitorio vanilloide 1 (TRPV1), a través de un metabolito intermedio del paracetamol, la N-acilfenolamina bioactiva (AM404) (Mallet et al., 2008).
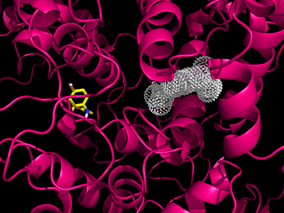
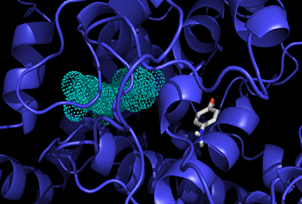
En esta investigación preliminar de Cuesta et. al. (2017), se propuso un estudio de la posible interacción del paracetamol y uno de sus metabolitos, el 4-aminofenol, con las enzimas encargadas de mediar el proceso inflamatorio, COX-1 y COX-2. Por medio del uso de técnicas computaciones, se propuso determinar la posibilidad que tienen estas moléculas para interaccionar e inhibir, hasta cierto punto, la actividad de estas dos ciclooxigenasas y así producir un efecto antiinflamatorio leve en el cuerpo. Para el estudio, la estructura del medicamento y su metabolito fueron optimizadas utilizando métodos mecánico cuánticos incluidos en el software Gaussian 09 (Frisch et al, 2009), mientras que la interacción de estas moléculas con las enzimas fue hecho con técnicas de acoplamiento molecular con el software autodock VINA. Las enzimas fueron obtenidas del Protein Data Bank (Berman et al., 2002). Para el caso de la COX-1, se utilizó la estructura cristalográfica de esta enzima unida al ibuprofeno, obtenida de una oveja a una resolución de 2,6 Å (código PDB: 1EQG) mientras que la para la COX-2, se usó la estructura cristalográfica de la enzima unida al medicamento celecoxib extraída de un ratón con una resolución de 2,4 Å (código PDB:3LN1).(Cuesta, Vela & Meneses, 2017)
Los resultados obtenidos que se presentan en la Tabla 3, muestran que las conformaciones con más afinidad para el paracetamol y 4-aminofenol no se encuentran en el sitio activo de la COX-1 y COX-2 (Figura 6). Se realizó el acoplamiento molecular con el sustrato natural de estas enzimas que es el ácido araquidónico y se encontraron valores de afinidad 1 kcal/mol mayores, lo que indicaría que tanto el paracetamol como el 4-aminofenol no ganarían en la competencia para ocupar el sitio activo COX durante un proceso inflamatorio. Esto sugiere que el mecanismo de acción principal del paracetamol o su metabolito no se da por inhibición de las Ciclooxigenasas (Cuesta, Vela, & Meneses, 2017). Este resultado concuerda con la literatura, donde se menciona una acción analgésica y antipirética, procesos que son mediados por otro sistema de enzimas (Jozwiak-Bebenista & Nowak, 2014). Adicionalmente, los efectos secundarios también son muy diferentes a los producidos por moléculas pertenecientes al grupo de los AINEs como el ibuprofeno. Estudios se continúan haciendo en esta área para determinar si el mecanismo de acción del paracetamol puede darse por la inhibición de los receptores cannabinoides lo que explicaría el efecto analgésico (Cuesta, Vela, & Meneses, 2017).
| Conformación | Afinidad COX-1 (kcal/mol) | Afinidad COX-2 (kcal/mol) | ||
| Paracetamol | 4-aminofenol | Paracetamol | 4-aminofenol | |
| 1 | -6,3 | -5,4 | -6,1 | -5,5 |
| 2 | -6,1 | -5,1 | -6,0 | -5,4 |
| 3 | -5,8 | -5,1 | -5,9 | -5,2 |
| 4 | -5,8 | -5,1 | -5,7 | -5,2 |
| 5 | -5,6 | -5,1 | -5,6 | -5,2 |
| 6 | -5,6 | -5,0 | -5,3 | -5,1 |
| 7 | -5,4 | -4,9 | -5,2 | -5,1 |
| 8 | -5,3 | -4,9 | -5,1 | -4,9 |
| 9 | -5,3 | -4,8 | -5,0 | -4,8 |
Nanoquímica
Las nanopartículas tienen propiedades ópticas, eléctricas y térmicas que son únicas (Saion, Gharibshahi, & Naghavi, 2013). Las aplicaciones de las nanopartículas van desde células fotovoltaicas a sensores biológicos y químicos. Las nanopartículas de plata han sido ampliamente estudiadas por sus aplicaciones biológicas en diagnóstico molecular, dispositivos fotónicos, sistemas nanométricos de liberación de fármacos, diagnóstico por imágenes y biosensores (Li et al., 2013; Romann, Wei, & Pileni, 2015). Pese a la gran aplicación de las nanopartículas, aún no se conoce a ciencia cierta las posibles consecuencias de su uso incluyendo su toxicidad y los posibles riesgos a la salud humana (Lewinski, Colvin, & Drezek, 2008). El modelamiento computacional es una herramienta útil para complementar los estudios experimentales. Con un estudio computacional, es factible simular interacciones bajo diferentes condiciones que no siempre es posible ser estudiado u observado en el laboratorio (You et al., 2012). El uso de computadoras en el área de la nanotecnología ha tenido resultados satisfactorios. Así, estudios teóricos sobre la afinidad de los iones de plata con el ADN a nivel molecular se han realizado para determinar la interacción de estos iones con la citosina y adenisona (Wu et al., 2012). Además, un análisis de reactividad química de moléculas biológicas en presencia de iones de plata se ha realizado para estudiar los orbitales moleculares y encontrar las especies más estables determinando, así, la reactividad (Martinez & Orrantia Borunda, 2016).
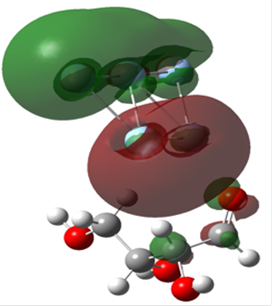
Siguiendo este enfoque, Fabara et.al. (2018) propuso estudiar la interacción entre las nanopartículas de plata con moléculas que se encuentran en la capa lipídica de la piel. Este estudio ha permitido tener indicios de cómo se comportan las nanoparticulas de plata cuando están en contacto con la piel. Para este estudio se propusieron cuatro sistemas, cada uno conteniendo diferentes átomos de plata (1, 3, 5 y 6). La energía de los sistemas fueron calculadas y sus estructuras optimizadas utilizando la teoría funcional de densidad implementada en el programa Gaussian 09 (Frisch et al, 2009). Como moléculas biológicas, para estudiar la interacción, sistemas orgánicos como glucosa, ácido esteárico, ácido palmítico y quercetina presentes en los recubrimientos de nanopartículas y en la piel fueron elegidas (Fabara, Cuesta, Pilaquinga, & Meneses, 2018).
Los resultados presentados en la Tabla 4 mostraron que los sistemas que tienen 5 átomos de plata fueron los que formaron los complejos más estables. Además, se realizó un estudio de orbitales moleculares, los cuales ayudaron a describir las interacciones mediante el estudio de los orbitales moleculares ocupados más externos (HOMO por sus siglas en ingles). Estos resultados mostraron que la densidad electrónica durante la interacción se encuentra alrededor de los átomos de plata (Figura 7). Simulaciones de dinámica molecular realizadas con el programa Abalone mostraron que las moléculas de plata se alejan de los sistemas orgánicos, sugiriendo la posibilidad de que estas nanopartículas tienen buenas propiedades de eliminación. (Fabara et.al., 2018).
| Sistema | ΔI (kcal/mol) | |||
| 1 Ag | 3 Ag | 5 Ag | 6 Ag | |
| Ácido esteárico | -2,79 | -18,58 | -8,51 | -75,47 |
| Ácido Palmítico | -3,41 | -18,65 | -8,53 | -75,43 |
| Quercetina | -0,81 | -24,44 | -12,53 | -80,16 |
| Glucosa | -26,50 | -- | -- | -- |
Péptidos bioactivos extraídos de anfibios
Las secreciones cutáneas de muchas ranas de la especie Anura contienen compuestos con actividad biológica que son muy interesantes para diseñar y desarrollar posibles nuevos fármacos (Conlon, 2012). Estas moléculas son parte del mecanismo de defensa del animal y del sistema de inmunidad innata, protegiendo a las ranas contra bacterias, hongos y protozoos (Conlon, 2012; Holthausen et al., 2017; Lacombe et al., 2015; Manzo et al., 2014; Scorciapino et al., 2013). Una gran parte de estas secreciones están compuestas por péptidos. Estos tienen un tamaño de 8 a 48 amino ácidos, una carga entre +2 y +6 a pH 7 debido a amino ácidos básicos como la lisina y al menos 50 % de amino ácidos hidrofóbicos (Conlon, 2012). Estudios han demostrado impresionantes propiedades de estos péptidos, que incluyen actividad contra virus, células cancerosas, antitumorales, antidiabéticos, antioxidantes, inhibición enzimática y efectos quimiostáticos (Holthausen et al., 2017; Marani et al., 2015). Actualmente, el uso de estos péptidos es limitado debido a su toxicidad desconocida hacia células mamíferas como los eritrocitos (Conlon, 2012).
En la literatura, no existe un mecanismo único por el cual los péptidos causen la muerte de las bacterias. Normalmente, su acción no implica interacción con un receptor específico, sino, una interacción no específica con la membrana celular de la bacteria que resulta en su permeabilización y desintegración (Conlon, 2012; Marani et al., 2015). Estas interacciones, a menudo complejas, pueden provocar que los péptidos se adhieran a la membrana y la permeabilicen, causando lisis celular. Esta es una de las razones por las que estos péptidos han funcionado contra bacterias resistentes a los antibióticos (Conlon, 2012; Stutz et al., 2017).
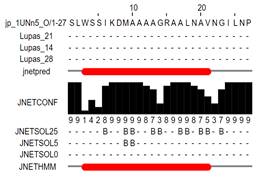
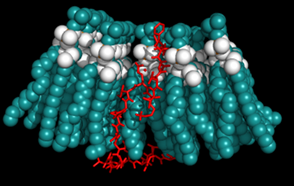
En esta investigación, se presentó un estudio computacional de cuatro Dermaseptinas extraídas del exudado de la piel de la rana Agalychnis spurrelli. Experimentalmente, estos péptidos fueron obtenidos, purificados y su secuencia de aminoácidos dilucidada usando diferentes técnicas. Adicionalmente, sus propiedades antimicrobianas contra bacterias Gram positivas, Gram negativas y antifúngicas fueron determinadas. Se encontró que DRS-SP2 tiene actividad contra E. coli, S. aureus y C. albicans. La DRS-SP3 y DRS-SP4 poseen actividad antimicrobiana contra E. coli y S. aureus, mientras que DRS-SP5 contra E. coli y C. albicans (Tabla 5). Con las secuencias en la mano, Cuesta et al., (2019a) y Cuesta et al., (2019b) realizaron un estudio computacional de las estructuras, obteniendo sus propiedades físico-químicas, estructura secundaria y su similitud con otros péptidos conocidos. Luego, se realizó un estudio de acoplamiento molecular de estos péptidos contra la membrana celular y varias enzimas que se sabe que son vitales para los organismos. Los resultados mostraron que las Dermaseptinas son péptidos catiónicos α-helicoidales (Figura 8) con un punto isoeléctrico superior a 9,70 y una carga positiva que va de +1 a +3 a pH fisiológico.


Al introducir estos péptidos en varias bases de datos, Cuesta et al., (2019b) determinó que su identidad en comparación con péptidos conocidos oscila entre 36 y 82 %, lo que significa que estas cuatro Dermaseptinas difieren a las encontradas en otras especies de ranas.
Los estudios de acoplamiento molecular realizados por Cuesta et al., (2019b) que se muestran en la Tabla 6, sugieren que el mecanismo de acción de este péptido no está dado por la inhibición enzimática, sino por lisis celular. Los resultados mostraron que el péptido interacciona con la membrana celular bacteriana atravesándola como se observa en la Figura 9. Resultados de la interacción con membrana de mamíferos mostró que en este caso, que los péptidos se mantienen en la superficie, lo que sugeriría una toxicidad baja en humanos (Cuesta et al., 2019a; Cuesta et al., 2019b). Estas son moléculas nuevas que tienen un gran potencial y que podrían estudiarse más a fondo, como punto de partida para fármacos antimicrobianos de última generación.
| Dermaseptina | Concentración Mínima inhibitoria | ||
| E. coli | S. aureus | C. albicans | |
| DRS-SP2 | 8 | 8 | 32 |
| DRS-SP3 | 128 | 512 | >512 |
| DRS-SP4 | 128 | 512 | >512 |
| DRS-SP5 | 256 | >512 | 1054 |
| Organismo | Enzima | Inhibidor conocido | Afinidad (kcal/mol) | ||||
| Inhibidor conocido | DRS-SP2 | DRS-SP3 | DRS-SP4 | DRS-SP5 | |||
| S. aureus | Proteína de unión de Penicilina G-acil 2ª | Ceftobiprole | -9,5 | -5,1 | -5,3 | -5,3 | -4,8 |
| Hidrolasa AmiA | Muramil tetrapeptido | -7,1 | -5,0 | -5,8 | -6,1 | -5,5 | |
| E. coli | ADN girasa B | ADP | -10,4 | -4,2 | -4,1 | -4,9 | -4,8 |
| Proteína de unión de Penicilina Transglicosilasa 1b | Moenomicina | -7,3 | -5,1 | -5,5 | -5,5 | -5,2 | |
| C. albicans | Exo-B-(1,3)-glucanasea | Castanospermina | -7,0 | -5,3 | -5,9 | -3,8 | -4,3 |
| Proteasa aspártica secretada | A70* | -7,7 | -6,1 | -6,4 | -6,4 | -6,5 | |
*A70: N-etil-N-[(4-metilpiperazin-1-il)carbonil]-D-fenilalanil-N-[(1S,2S,4R)-4-(butilcarbamoil)-1-(ciclohexilmetil)-2-hidroxi-5-metilhexil]-L-norleucinamida

CONCLUSIÓN
La química computacional centra sus bases en la fisicoquímica y el comportamiento de los átomos en un sistema dinámico. Desde sus inicios en los años 1930, la química computacional nunca ha sido tan importante como en los últimos 10 años. Los avances de la tecnología a pasos agigantados, ha permitido que cada vez los nuevos métodos y técnicas computacionales sean de mayor utilidad, reproduciendo con un bajo porcentaje de error resultados obtenidos experimentalmente. Así, herramientas computacionales consiguen predecir la reactividad de nitroarenos basado en descriptores como la electrofilia global. Adicionalmente, se puede tener un mejor entendimiento del mecanismo de reacción, como el de la producción de VAD, con lo que se pueden diseñar mecanismos más eficientes para la síntesis de este compuesto o usar el mecanismo para obtener compuestos diferentes. En la rama de la farmacología, se puede llegar a entender a nivel molecular las interacciones que una molécula, como las Dermaseptinas, el paracetamol o nanopartículas de plata, tiene con distintas enzimas o biomoléculas, pudiendo así explicar las propiedades bactericidas, analgésicas y antiinflamatorias, que muestran estos compuestos experimentalmente.
Hoy en día, las técnicas computacionales han alcanzado un nivel de eficiencia tan alto, reduciendo el proceso de descubrimiento de un medicamento de 10 a 4 años con moléculas mucho más seguras y eficaces. De igual manera, la inteligencia artificial aplicada a esta rama ha permitido tener rutas de síntesis súper eficientes, comparadas a las que serían obtenidas por químicos orgánicos con más de 20 años de experiencia en el campo. Y así, investigadores alrededor del mundo pueden abordar una infinidad de problemas, pudiendo sentar las bases para procesos químicos más eficientes, dar luz en mecanismos de acción de fármacos desconocidos y caracterizar nuevas moléculas que se pueden convertir en los nuevos fármacos, sin tener que invertir grandes sumas de dinero en equipos reactivos e infraestructura en una etapa inicial.
LISTA DE REFERENCIAS
Bell, S., Dines, T. J., Chowdhry, B. Z., & Withnall, R. (2007). Computational chemistry using modern electronic structure methods. Journal of Chemical Education, 84(8), 1364–1370. https://doi.org/10.1021/ed084p1364
Berman, H. M., Battistuz, T., Bhat, T. N., Bluhm, W. F., Bourne, P. E., Burkhardt, K., … Zardecki, C. (2002). The protein data bank. Acta Crystallographica Section D: Biological Crystallography, 28, 235–242. https://doi.org/10.1107/S0907444902003451
Botting, R., & Ayoub, S. S. (2005). COX-3 and the mechanism of action of paracetamol/acetaminophen. Prostaglandins Leukotrienes and Essential Fatty Acids, 72, 85–87. https://doi.org/10.1016/j.plefa.2004.10.005
Conlon, J. M. (2012). The potential of frog skin antimicrobial peptides for development into therapeutically valuable anti-infective agents. In ACS Symposium Series (Vol. 1095, pp. 47–60). American Chemical Society. https://doi.org/10.1021/bk-2012-1095.ch003
Cuesta, S.; Arias, J.; Gallegos, F.; Alzate-Morales, J.; Meneses, L. (2018). On the Reaction Mechanism of the 3,4-Dimethoxybenzaldehyde Formation from 1-(3′,4′-Dimethoxyphenyl)Propene. Molecules, 23(2), E412. https://doi.org/10.3390/molecules23020412
Cuesta H., S., Arias de P., J., Gallegos P., F., Proaño B., C., Blasco-Zúñiga, A., Rivera I., M., & Meneses O., L. (2019a). Modelamiento molecular de la dermaseptina SP2 extraída de Agalychnis spurrelli. InfoANALÍTICA, 7(1), 41. https://doi.org/10.26807/ia.v7i1.95
Cuesta, S., Gallegos, F., Arias, J., Pilaquinga, F., Blasco-Zúñiga, A., Proaño-Bolaños, C., … Meneses, L. (2019b). Molecular modeling of four Dermaseptin-related peptides of the gliding tree frog Agalychnis spurrelli. Journal of Molecular Modeling, 25(9). https://doi.org/10.1007/s00894-019-4141-1
Cuesta, S., Vela, C., & Meneses, L. (2017). Modelación molecular de la interacción del paracetamol y el 4-aminofenol con las enzimas Ciclooxigenasa 1 y 2. Revista Ecuatoriana de Medicina y Ciencias Biológicas. https://doi.org/10.26807/remcb.v38i2.546
Dashtban, M., Schraft, H., Syed, T. A., & Qin, W. (2010). Fungal biodegradation and enzymatic modification of lignin. International Journal of Biochemistry and Molecular Biology.
Egwim, E. C., Kabiru, A. Y., & Tola, A. J. (2015). Partial characterization of lignin peroxidase expressed by bacterial and fungal isolates from termite gut. Biokemistri, 27(1), 33–38. Retrieved from http://www.bioline.org.br/bk
Fabara, A., Cuesta, S., Pilaquinga, F., & Meneses, L. (2018). Computational Modeling of the Interaction of Silver Nanoparticles with the Lipid Layer of the Skin. Journal of Nanotechnology, 2018. https://doi.org/10.1155/2018/4927017
Foresman, J. B. (2001). Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems By David Young (Cytoclonal Pharmaceutics Inc.). Wiley-Interscience: New York. 2001. xxvi + 382 pp. $69.95. ISBN: 0-471-33368-9. Journal of the American Chemical Society, 123(41), 10142–10143. https://doi.org/10.1021/ja015246y
Fourches, D., Muratov, E., & Tropsha, A. (2010, July 26). Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. Journal of Chemical Information and Modeling. https://doi.org/10.1021/ci100176x
Frisch, M. J.; Trucks, G.W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.;Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenber, D. J. (2009). Gaussian 09. Gaussian, Inc. Wallingford CT.
Graham, G. G., Davies, M. J., Day, R. O., Mohamudally, A., & Scott, K. F. (2013). The modern pharmacology of paracetamol: Therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacology, 21, 201–232. https://doi.org/10.1007/s10787-013-0172-x
Graham, G. G., & Scott, K. F. (2005). Mechanism of action of paracetamol. American Journal of Therapeutics, 12, 46–55. https://doi.org/10.1097/00045391-200501000-00008
Harada, A., Sasaki, K., & Kaneta, T. (2016). Direct determination of lignin peroxidase released from Phanerochaete chrysosporium by in-capillary enzyme assay using micellar electrokinetic chromatography. Journal of Chromatography A, 1440, 145–149. https://doi.org/10.1016/j.chroma.2016.02.062
Hassan, M., Brown, R. D., Varma-O’Brien, S., & Rogers, D. (2006, August). Cheminformatics analysis and learning in a data pipelining environment. Molecular Diversity. https://doi.org/10.1007/s11030-006-9041-5
Holthausen, D. J., Lee, S. H., Kumar, V. T., Bouvier, N. M., Krammer, F., Ellebedy, A. H., … Jacob, J. (2017). An Amphibian Host Defense Peptide Is Virucidal for Human H1 Hemagglutinin-Bearing Influenza Viruses. Immunity, 46(4), 587–595. https://doi.org/10.1016/j.immuni.2017.03.018
Holtje, H. D., Folkers, G., & Luzar, A. (1998). Molecular Modeling, Basic Principles and Applications. Computers in Physics, 12(1), 41. https://doi.org/10.1063/1.168645
Jozwiak-Bebenista, M., & Nowak, J. Z. (2014). Paracetamol: Mechanism of action, applications and safety concern. Acta Poloniae Pharmaceutica - Drug Research, 71(1), 11–23.
Karlström, G., Lindh, R., Malmqvist, P. Å., Roos, B. O., Ryde, U., Veryazov, V., … Seijo, L. (2003). MOLCAS: A program package for computational chemistry. In Computational Materials Science (Vol. 28, pp. 222–239). https://doi.org/10.1016/S0927-0256(03)00109-5
Khindaria, A., Yamazaki, I., & Aust, S. D. (1995). Veratryl Alcohol Oxidation by Lignin Peroxidase. Biochemistry, 34(51), 16860–16869. https://doi.org/10.1021/bi00051a037
Kis, B., Snipes, A., Bari, F., & Busija, D. W. (2004). Regional distribution of cyclooxygenase-3 mRNA in the rat central nervous system. Molecular Brain Research, 126, 78–80. https://doi.org/10.1016/j.molbrainres.2004.03.015
Kis, B., Snipes, J. A., & Busija, D. W. (2005). Acetaminophen and the cyclooxygenase-3 puzzle: Sorting out facts, fictions, and uncertainties. Journal of Pharmacology and Experimental Therapeutics, 315, 1–7. https://doi.org/10.1124/jpet.105.085431
Kudo, S., Harada, A., Kubota, H., Sasaki, K., & Kaneta, T. (2017). Simultaneous Determination of Manganese Peroxidase and Lignin Peroxidase by Capillary Electrophoresis Enzyme Assays. ACS Omega, 2(10), 7329–7333. https://doi.org/10.1021/acsomega.7b00998
Lacombe, C., Piesse, C., Sagan, S., Combadière, C., Rosenstein, Y., & Auvynet, C. (2015). Pachymodulin, a new functional formyl peptide receptor 2 peptidic ligand isolated from frog skin has janus-like immunomodulatory capacities. Journal of Medicinal Chemistry, 58(3), 1089–1099. https://doi.org/10.1021/jm501018q
Lewars, E. G., & Lewars, E. G. (2016a). An Outline of What Computational Chemistry Is All About. In Computational Chemistry (pp. 1–8). Springer International Publishing. https://doi.org/10.1007/978-3-319-30916-3_1
Lewars, E. G., & Lewars, E. G. (2016b). Introduction to Quantum Mechanics in Computational Chemistry. In Computational Chemistry (pp. 101–191). Springer International Publishing. https://doi.org/10.1007/978-3-319-30916-3_4
Lewars, E. G., & Lewars, E. G. (2016c). Molecular Mechanics. In Computational Chemistry (pp. 51–99). Springer International Publishing. https://doi.org/10.1007/978-3-319-30916-3_3
Lewinski, N., Colvin, V., & Drezek, R. (2008, January). Cytotoxicity of nanopartides. Small. https://doi.org/10.1002/smll.200700595
Li, R., Chen, R., Chen, P., Wen, Y., Ke, P. C., & Cho, S. S. (2013). Computational and experimental characterizations of silver nanoparticle-apolipoprotein biocorona. Journal of Physical Chemistry B, 117(43), 13451–13456. https://doi.org/10.1021/jp4061158
MacDonald, J., Goacher, R. E., Abou-Zaid, M., & Master, E. R. (2016). Comparative analysis of lignin peroxidase and manganese peroxidase activity on coniferous and deciduous wood using ToF-SIMS. Applied Microbiology and Biotechnology, 100(18), 8013–8020. https://doi.org/10.1007/s00253-016-7560-2
Mąkosza, M. (2010). Nucleophilic substitution of hydrogen in electron-deficient arenes, a general process of great practical value. Chemical Society Reviews, 39(8), 2855–2868. https://doi.org/10.1039/b822559c
Ma̧kosza, M., Lemek, T., Kwast, A., & Terrier, F. (2002). Elucidation of the vicarious nucleophilic substitution of hydrogen mechanism via studies of competition between substitution of hydrogen, deuterium, and fluorine. Journal of Organic Chemistry, 67(2), 394–400. https://doi.org/10.1021/jo010590z
Ma̧kosza, M., & Wojciechowski, K. (2004). Nucleophilic substitution of hydrogen in heterocyclic chemistry. Chemical Reviews, 104(5), 2631–2666. https://doi.org/10.1021/cr020086+
Mallet, C., Daulhac, L., Bonnefont, J., Ledent, C., Etienne, M., Chapuy, E., … Eschalier, A. (2008). Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain, 139, 190–200. https://doi.org/10.1016/j.pain.2008.03.030
Manzo, G., Casu, M., Rinaldi, A. C., Montaldo, N. P., Luganini, A., Gribaudo, G., & Scorciapino, M. A. (2014). Folded structure and insertion depth of the frog-skin antimicrobial peptide esculentin-1b(1-18) in the presence of differently charged membrane-mimicking micelles. Journal of Natural Products, 77(11), 2410–2417. https://doi.org/10.1021/np5004406
Marani, M. M., Dourado, F. S., Quelemes, P. V., De Araujo, A. R., Perfeito, M. L. G., Barbosa, E. A., … Leite, J. R. S. A. (2015). Characterization and Biological Activities of Ocellatin Peptides from the Skin Secretion of the Frog Leptodactylus pustulatus. Journal of Natural Products, 78(7), 1495–1504. https://doi.org/10.1021/np500907t
Martinez, L.-L. L., & Orrantia Borunda, E. (2016). DFT Chemical Reactivity Analysis of Biological Molecules in the Presence of Silver Ion. Organic Chemistry: Current Research, 04(04). https://doi.org/10.4172/2161-0401.1000153
Meneses, L., Morocho, S., Castellanos, A., & Cuesta, S. (2017). Computational study of vicarious nucleophilic substitution reactions. Journal of Molecular Modeling, 23(10). https://doi.org/10.1007/s00894-017-3464-z
Ottani, A., Leone, S., Sandrini, M., Ferrari, A., & Bertolini, A. (2006). The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. European Journal of Pharmacology, 531, 280–281. https://doi.org/10.1016/j.ejphar.2005.12.015
Parr, R. G. (1980). Density Functional Theory of Atoms and Molecules. In Horizons of Quantum Chemistry. https://doi.org/10.1007/978-94-009-9027-2_2
Parr, R. G., Szentpály, L. V., & Liu, S. (1999). Electrophilicity index. Journal of the American Chemical Society, 121(9), 1922–1924. https://doi.org/10.1021/ja983494x
Parr, R. G., & Yang, W. (1984). Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. Journal of the American Chemical Society, 106(14), 4049–4050. https://doi.org/10.1021/ja00326a036
Patz, M., Mayr, H., Maruta, J., & Fukuzumi, S. (1995). Reactions of Carbocations with π Nucleophiles: Polar Mechanism and No Outer Sphere Electron Transfer. Angewandte Chemie International Edition in English, 34(11), 1225–1227. https://doi.org/10.1002/anie.199512251
Pérez, P., Domingo, L. R., Aurell, M. J., & Contreras, R. (2003). Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1,3-dipolar cycloaddition reactions. Tetrahedron, 59(17), 3117–3125. https://doi.org/10.1016/S0040-4020(03)00374-0
Rai, N., Yadav, M., & Singh Yadav, H. (2016). Enzymatic Characterisation of Lignin Peroxidase from Luffa aegyptiaca Fruit Juice. American Journal of Plant Sciences, 07(03), 649–656. https://doi.org/10.4236/ajps.2016.73057
Romann, J., Wei, J., & Pileni, M. P. (2015). Computational matching of surface plasmon resonance: Interactions between silver nanoparticles and ligands. Journal of Physical Chemistry C, 119(20), 11094–11099. https://doi.org/10.1021/jp511859p
Saion, E., Gharibshahi, E., & Naghavi, K. (2013). Size-controlled and optical properties of monodispersed silver nanoparticles synthesized by the radiolytic reduction method. International Journal of Molecular Sciences, 14(4), 7880–7896. https://doi.org/10.3390/ijms14047880
Schindele, C., Houk, K. N., & Mayr, H. (2002). Relationships between carbocation stabilities and electrophilic reactivity parameters, E: Quantum mechanical studies of benzhydryl cation structures and stabilities. Journal of the American Chemical Society, 124(37), 11208–11214. https://doi.org/10.1021/ja020617b
Scorciapino, M. A., Manzo, G., Rinaldi, A. C., Sanna, R., Casu, M., Pantic, J. M., … Conlon, J. M. (2013). Conformational analysis of the frog skin peptide, plasticin-L1, and its effects on production of proinflammatory cytokines by macrophages. Biochemistry, 52(41), 7231–7241. https://doi.org/10.1021/bi4008287
Shah, M. M., Grover, T. A., Barr, D. P., & Aust, S. D. (1992). On the mechanism of inhibition of the veratryl alcohol oxidase activity of lignin peroxidase H2 by EDTA. Journal of Biological Chemistry, 267(30), 21564–21569.
Snipes, J. A., Kis, B., Shelness, G. S., Hewett, J. A., & Busija, D. W. (2005). Cloning and characterization of cyclooxygenase-1b (putative cyclooxygenase-3) in rat. Journal of Pharmacology and Experimental Therapeutics, 313(2), 668–676. https://doi.org/10.1124/jpet.104.079533
Stutz, K., Müller, A. T., Hiss, J. A., Schneider, P., Blatter, M., Pfeiffer, B., … Schneider, G. (2017). Peptide-Membrane Interaction between Targeting and Lysis. ACS Chemical Biology, 12(9), 2254–2259. https://doi.org/10.1021/acschembio.7b00504
Ten Have, R., Franssen, M. C. R., & Field, J. A. (2015). Lignin peroxidase initiates O2-dependent self-propagating chemical reactions which accelerate the consumption of 1-(3′,4′-dimethoxyphenyl)propene. Biochemical Journal, 347(2), 585–591. https://doi.org/10.1042/bj3470585
Vollhardt, P., & Schore, N. (2014). Organic Chemistry. Organic Chemistry. Macmillan Learning. https://doi.org/10.1007/978-1-319-19197-9
Winiarski, J., & Makosza, M. (1987). Vicarious Nucleophilic Substitution of Hydrogen. Accounts of Chemical Research, 20(8), 282–289. https://doi.org/10.1021/ar00140a003
Wu, J., Fu, Y., He, Z., Han, Y., Zheng, L., Zhang, J., & Li, W. (2012). Growth mechanisms of fluorescent silver clusters regulated by polymorphic DNA templates: A DFT study. Journal of Physical Chemistry B, 116(5), 1655–1665. https://doi.org/10.1021/jp206251v
You, C., Han, C., Wang, X., Zheng, Y., Li, Q., Hu, X., & Sun, H. (2012, September). The progress of silver nanoparticles in the antibacterial mechanism, clinical application and cytotoxicity. Molecular Biology Reports. https://doi.org/10.1007/s11033-012-1792-8
Zhao, Y., & Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts, 120(1–3), 215–241. https://doi.org/10.1007/s00214-007-0310-x

