INTRODUCCIÓN
La patología motriz no progresiva de la
infancia engloba a diferentes entidades que son su causa o que con frecuencia
se asocian a ella, unas veces ocurre por daño directo de neuronas o sus fibras
que conforman las vías motoras y/o sus conexiones a nivel central (parálisis
cerebral de cualquier etiología), por alteración genética de la matriz
extracelular de donde la célula toma elementos para su propio funcionamiento y
sobrevivencia (dismetabolias electrolíticas, proteicas, enzimáticas, etc.), por
combinaciones de las anteriores o por otros mecanismos no bien establecidos. En
todo caso, esa disfunción motora es un estado neurológico secuelar y no una
enfermedad en sí, es una discapacidad física, puede complicarse o asociarse con
alguna alteración progresiva u otras neurológicas, dando la errónea apreciación
de que está empeorando: en estos casos lo que hay son nuevas lesiones por la entidad
nosológica agregada. Muchas de las actividades de la vida diaria tiene un
componente motriz importante: el habla, la respiración, la excreción endócrina,
el peristaltismo enteral, la lectura, uso de extremidades, etc.: todas ellas,
en grado variable, disfuncionan cuando hay daño de la expresada vía por acción
o consecuencia de malformaciones genéticas o congénitas, hipoxia, isquemia,
infección, trauma directo, o acción de agentes
físicos y químicos. El conocer si está presente alguna alteración metabólica es
importante porque a las medidas terapéuticas eminentemente físicas y pedagógicas
con que se tratan estas secuelas, se agregarían las nutricionales y
farmacológicas apropiadas(1-3).
Los defectos en el metabolismo de
aminoácidos o de ácidos orgánicos ocasionan sintomatología en parte motora, a
causa de trastornos por excesiva presencia intracelular de sus metabolitos dada la falla
enzimática. Están descritas más de 800 de estas patologías monogénicas, con
herencia autosómica recesiva en su mayoría, que se expresan por ausencia o
falla de uno o varios genes o de su transportador postranscripcional. Mutaciones
genéticas producen síntesis de proteínas anormales que ocasionan fisiología
disfuncional, con acúmulo de sustancias intermedias o finales anormales, las
cuales tendrán efectos según la toxicidad de las mismas; otras mutaciones
genéticas provocan uso de vías metabólicas no apropiadas, las cuales forman sustancias
intermedias o productos finales anormales, que, al acumularse, devienen en toxicidad(4-6).
El
objetivo de este estudio fue determinar, en una población de niños y
adolescentes con Parálisis Cerebral (PC) y Errores Innatos Metabólicos (EIM), cuáles
son los diversos tipos de EIM en esa combinación de noxas –con una prevalencia de
1-2,5 por mil nacidos vivos.
MÉTODOS
Estudio retrospectivo descriptivo interpretativo
en el cual se revisaron 2.000 Historias Clínicas del archivo del Centro de
Parálisis Cerebral de Caracas (institución pública venezolana) de menores de 18
años, atendidos entre los años 1998 y 2018, con diagnóstico (Dx.) de PC de todo
tipo y Dx agregado de EIM, confirmado por la clínica y el laboratorio.
RESULTADOS
De las 2000 historias clínicas de
infantes con PC diagnosticadas entre 1988 y 2018, el examen clínico y las pruebas de laboratorio
permitieron seleccionar 174 historias clínicas con EIM como Dx agregado, como
se especifica en la Tabla 1.
Tabla 1.
Errores innatos metabólicos en 174 niños con parálisis cerebral.
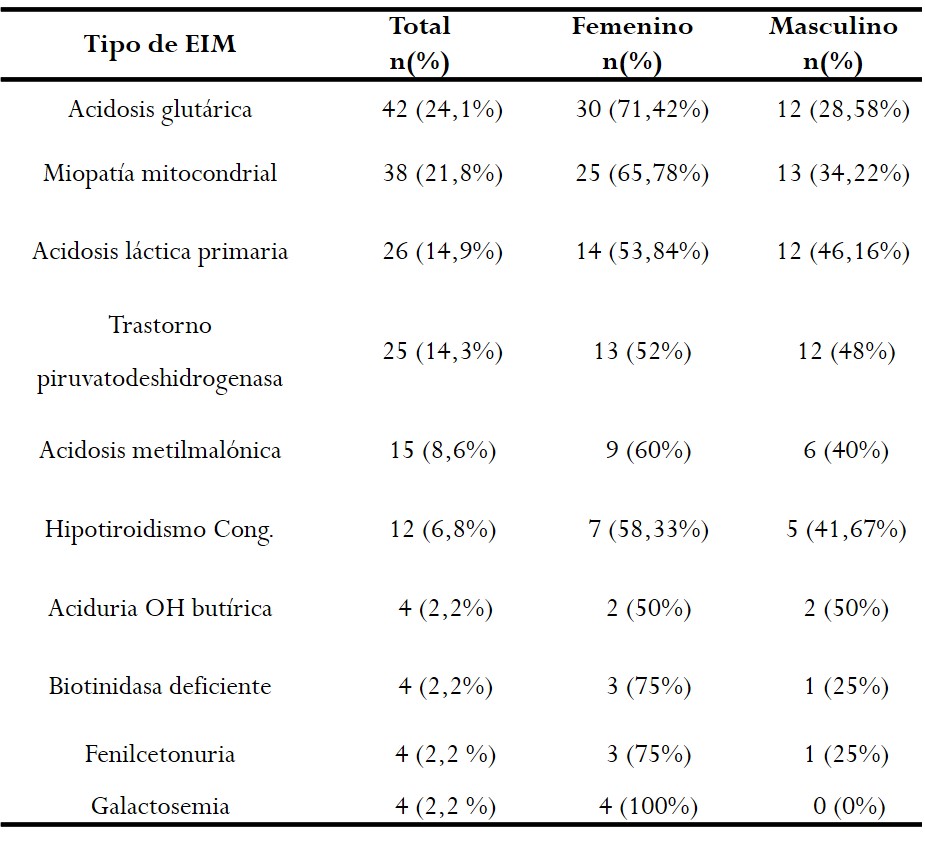 Fuente: Archivo
de Historias Médicas, Centro de Parálisis Cerebral (Casuística propia) EEI: errores innatos
metabólicos.
Fuente: Archivo
de Historias Médicas, Centro de Parálisis Cerebral (Casuística propia) EEI: errores innatos
metabólicos.
DISCUSIÓN
En los EIM de aminoácidos y/o de ácidos
orgánicos los organelos celulares que con más frecuencia están afectados y
causan trastornos degenerativos, metabólicos o ambos son: mitocondrias,
lisosomas y peroxisomas; dado que deficiencias enzimáticas impiden que los
nutrientes sean transformados en energía, en cualquiera de esos 3 casos se
acumularan sustancias biológicas dentro de las células de manera anormal, o
productos intermedios de ellas, por ello el daño es doble: acúmulo anormal y
falla en la producción energética al no ser metabolizadas. Raramente cualquier otro organelo puede estar
afectado como para expresar un error innato, mostrando –como los otros- algunas
alteraciones en resonancias magnéticas cerebrales: refringencias bilaterales,
sobre todo en núcleos lenticulares y tálamo.
Las mitocondrias -que se cree es una asociación protistal con una célula compleja-, tienen su propio ADN, diferente al del núcleo celular, poseen su propio ritmo replicativo; en la concepción, las células hijas luego de dividirse el huevo o cigoto, solo reciben mitocondrias maternas (recordar que al óvulo engullir al espermatozoide lo disuelve y solo su material cromosómico fecundante es lo que queda de él): por esa razón las patologías de estos organelos por vía paterna no existen(7). La respiración celular (fosforilación oxidativa) produce el adenosin trifosfato (ATP) que es la fuente energética celular(8). Los lisosomas son organelos derivados del aparato de Golgi contentivos de hidrolasas sintetizadas en el retículo endoplásmico rugoso, estas enzimas almacenadas, al liberarse pueden digerir la totalidad de la célula(9). En cuanto a los peroxisomas, por gemación deriva este organelo del retículo endoplásmico liso, almacena diversas enzimas (catalasas, peroxidasas) que actúan en el metabolismo lipídico y de aminoácidos(10).
En las alteraciones mitocondriales no se producen niveles de ATP fisiológicamente adecuados, por eso hay en sangre, Líquido Céfalorraquídeo (LCR) y músculo, cantidades excesivas de alanina, lactato y piruvato(11); en alteraciones lisosomales, el déficit de hidrolasas hace que se depositen excesivamente mucopolisacáridos, lípidos y/o carbohidratos dentro de la célula, sin autofagocitarse: entonces se producirán leucodistrofias y mucopolisacaridosis; en éste último grupo de patologías glucídicas no hay un adecuado catabolismo lisosomal de tres glucosaminoglicanos: heparán sulfato, dermatán sulfato y/o keratán sulfato, lo cual impide la excreción de ellos por vía urinaria y las consecuencias serán: dismorfias óseas, alteraciones visuales progresivas y regresión cognoscitiva(12).
En alteraciones de los peroxisomas, la deficiencia de la enzima acetilCoA sintetasa (necesaria para mielinización) hace que los ácidos grasos se acumulen, produciéndose leucodistrofia (no se recupera la mielina lesionada en sustancia blanca cerebral), displasia cortical (polimicrogiria y paquigiria), cataratas, hipotonía al principio; cuando es progresiva, la sobrevida esperada es hasta el final de la primera década de vida(13).
Las deficiencias enzimáticas o enzimas defectuosas impiden el metabolismo normal de glúcidos, lípidos y prótidos, no permiten que los nutrientes sean transformados en energía por falta de parte de vía metabólica, y se acumulan sustancias biológicas intracelulares y/o productos intermedios, es decir, hay un doble mecanismo de daño o falla doble: por el acúmulo anormal y por falla en la producción energética. La resultante: disfunción motriz –voluntaria e involuntaria- y en muchos casos, alteraciones de funciones ejecutivas. El origen de todo esto, más que errores al momento de la regeneración diploide del óvulo fecundado, es por mutación, aunque un gen mutado no necesariamente provoca trastorno pues está el otro alelo (no mutado), y están el medio ambiente y el microbioma; finalmente, pocos EIM tienen un fenotipo típico(14).
Hay asociaciones entre EIM y PC hipóxica-isquémica, es difícil saber en estos casos mixtos si son los primeros los que ocasionan la alteración del movimiento o si traen como consecuencia las complicaciones descritas al principio que lesionan los centros y vías motrices centrales propias de la PC; en todo caso, en Medicina, la asociación no necesariamente es igual a etiología.(15)
Clínicamente, en los EIM lo más evidente al principio son las fallas motrices, a pesar de que se afecta todo el organismo; al nacer muchos se observan muy afectados como sepsis neonatal, la motricidad patológica es inespecífica -mermada por hiper o hipotonía según la deficiencia presente-, presencia de movimientos involuntarios en extremidades y respuesta disminuida ante estímulos ambientales. Más adelante se le adicionan alteraciones en control de peristaltismo manifestadas como reflujo gastroesofágico y emesis frecuentes; tardíamente, cuando se logra la marcha ésta es atáxica. Los otros componentes signo sintomatológicos más comunes son: retardo mental, convulsiones, deformidades óseas y alteraciones musculares o en órganos internos.
Los hallazgos que apunten a EIM dependen de la edad, en recién nacidos: convulsiones, apneas, emesis, intolerancias alimentarias y deterioro del estado de conciencia; en las semanas siguientes puede ser la misma situación o empeoramiento con cambios nutricionales, hiperlactacidemia, hiperamonemia o cetoacidosis; retardo motor e infecciones frecuentes. Puede haber períodos intercríticos asintomáticos; luego se acentúan intolerancias alimentarias, se evidencia retardo psicomotor y dismorfias, alteraciones moderadas de órganos vitales, y ya en escolaridad: dificultades en aprendizaje, retardo mental y/o del crecimiento(16).
Por todas estas razones es que el laboratorio general neonatal y de primera infancia incluye hematología, gasometría y electrolitos séricos, química sanguínea básica, ácido cetónico (DPNH) y cetonuria, amonio, ácido láctico en sangre, aminoácidos en sangre y cromatografía de ácidos orgánicos en orina, CPK total y fraccionada, tirosinemia, y además de las pruebas propias para detección de alteraciones en aminoácidos y ácidos orgánicos, tanto en sangre como en orina(17). Aunque pocas veces se cuenta con imágenes de resonancia cerebral, con frecuencia se ve hiperintensidad en corona radiada, striatum o periventricular(18).
Las medidas terapéuticas de urgencia buscan corregir desequilibrios y pueden incluir hemodiálisis SOS; las de mantenimiento son básicamente restricciones dietéticas según el caso y uso de fármacos, L-carnitina en todos los casos, las más específicas
dependen de si es necesario aplicar terapia farmacológica de inhibición de sustrato o de reemplazo(19).
En la acidosis/aciduria glutárica hay deficiencia de GlutarilCoA deshidrogenasa para catabolizar a los aminoácidos: triptófano, lisina e hidroxilisina; se degeneran los núcleos estriados basales telencefálicos, y aparecen retardo motriz y distonías. Se indica dieta evitando esos aminoácidos y se agrega L carnitina. La Aciduria glutárica tipo II difiere de la tipo I en que habiendo dos proteínas defectuosas: flavoproteína de transferencia de electrones (FTE), y FTE-ubiquinona oxidoreductasa, se interrumpen secuencias metabólicas en la cadena respiratoria por lo cual tanto el anabolismo como el catabolismo de aminoácidos y ácidos grasos se alteran, acumulándose el ácido glutárico de manera anormal, por lo cual la clínica es de acidosis común(20).
La encéfalomiopatía mitocondrial: ocasiona baja energía, exceso de radicales libres y también acidosis láctica, por eso sus efectos son sistémicos. Habrá debilidad muscular e intolerancia al ejercicio, ptosis palpebral asimétrica y estrabismo(21).
En la acidosis láctica primaria o congénita hay un trastorno de la vía metabólica de la alanina y de la glucólisis, la dismetabolia es con el piruvato, acumulándose lactato en sangre y LCR, el resultado es espasticidad y a veces coreoatetosis, diarrea frecuente, trastornos cutáneos y acidosis persistente con hiperventilación compensatoria. Las medidas habituales son hidratación y uso de bicarbonato vía endovenosa(22).
Trastornos en la enzima piruvato-deshidrogenasa o acidosis láctica por deficiencia de piruvato deshidrogenasa: tres enzimas de ese complejo transforman al piruvato en Acetil Coa (mediante decarboxilacion del piruvato), ese Acetil CoA es usada en el ciclo del citrato para la respiración celular; aparecen anomalías cerebrales. Es uno de los EIM más estudiados, los genes que mutan son: PDHA1, PDHB, PDHX PDP1 y DLAT. En estos pacientes hay retardo mental, acidosis metabólica, alaninuria, hiperlactacidemia, incoordinación muscular y empeoran con stress o al ingerir carbohidratos. El crecimiento prenatal es lento, y aparece acidosis neonatal. Se utilizan L-carnitina, tiamina y dieta cetogénica(23).
En la aciduria/acidemia metilmalónica pueden darse alguna de tres posibilidades: deficiencia de la enzima metilmalonil1-CoA epimerasa, de la enzima metilmalonil CoA mutasa o déficit en el transporte del cofactor adenosil cobalamina; por tal razón se acumula ácido metilmalónico; es una acidemia orgánica en la cual hay hiperamonemia, hiperlactacidemia y acidosis metilmalónica; los recién nacidos son normales al iniciar ingesta de nutrientes, entonces aparecen reflujo gastroesofágico y emesis, movimientos espontáneos anormales, retardo psicomotor e hipotonía. Es útil en ellos no guardar ayuno, vigilar niveles de cuerpos cetónicos y el uso de L-carnitina y vitamina B-12(24). No confundir con hiperamonemia primaria debido a deficiencias en enzimas que metabolizan al amonio, lo cual es mucho más grave porque empeora con la ingesta proteica y se producen rápidas lesiones en el sistema nervioso central (en resonancia cerebral: núcleo caudado con alta señal y atrofia cerebral); la sintomatología de hiperamonemia neonatal es: hipotonía, emesis, debilidad hasta para succionar, depresión del estado de conciencia y crisis comiciales (en estos casos la cromatografía de ácidos orgánicos en orina es normal), no habrá la acidosis de la metilmalónica y se alteran otros valores: ornitína, citrulina, arginina y ácido orótico(25).
Aciduria hidroxibutírica: una mutación en el gen SSADH (cromosoma 6p22) provoca falla en la síntesis de la enzima semialdehido succínico deshidrogenasa para metabolizar al ácido gammaaminobutírico GABA (neurotransmisor inhibidor cerebral), por eso se acumulará y eliminará en exceso el ácido OH- butírico, detectándose su aumento en orina, sangre y LCR. Clínicamente habrá retardo mental severo (algunos afortunados solo retraso psicomotor y retardo en desarrollo del lenguaje), hipotonía, hiporreflexia y ataxia, luego aparecen las convulsiones de difícil control, déficit de atención y agresividad. En resonancias aparece refringencia palidal y de núcleos cerebelosos. Su tratamiento es sintomático(26).
Deficiencia de biotinidasa: la enzima biotinidasa (codificada en el cromosoma 3p25) permite el procesamiento de la biotina dietaria, el uso y reciclaje de esta vitamina hidrosoluble del complejo B que participa en reacciones mediadas por carboxilasas para gluconeogénesis, catabolismo de aminoácidos y síntesis de ácidos grasos; las consecuencias serán: hipotonía, retardo psicomotor, ataxia, disminución en funcionalismo opto-acústico, convulsiones, disfunción acústica y trastornos cutáneos; hiperamonemia leve, acidosis sérica y aciduria. Se indican suplementos de biotina(27).
En la galactosemia se imposibilita el paso de galactosa a glucosa; cursa con insuficiencia ovárica, renal, hepática y cataratas, alteraciones del lenguaje y discinesias a nivel motriz; se recomienda dieta sin galactosa, aun así, la mejoría cognoscitiva es escasa(28). En el
hipotiroidismo congénito, un 20% son por EIM y no por defecto primario de la
glándula tiroidea, cursa con retardo mental; en laboratorio se reportan valores
bajos en el perfil tiroideo. Se indica reemplazo hormonal con L-tirosina(29). Otros
errores metabólicos de origen genético con frecuencia reportados como la fenilcetonuria
(hipo o normotirosinemia con hiperfenilalaninemia desde el nacimiento debido a
falla de la enzima fenilalanina hidroxilasa), adrenoleucodistrofia, tirosinemia
genética y acidemia isovalérica, no se encontraron en nuestro estudio de PC con
EIM(30): ¿fallas en diagnóstico?, ¿ausencia real de estas patologías?,
¿o por inexistencia de instituciones que gratuitamente procesen esto de manera
oportuna? En la fenilcetonuria hay cognición baja, hiperactividad, motricidad
anómala, conducta disruptiva; se indica dieta sin fenilalanina.
En nuestra casuística, se observa coincidencia entre nuestros resultados y los reportados en la literatura en lo relativo a frecuencia, predominancias por sexo y a no poder establecer con seguridad si el trastorno motriz en estos casos mixtos fue causado por hipoxia, infección, o por un EIM: cualquiera de los tres puede generar una secuencia en cascada con impacto en los centros motrices de los pacientes por vía de falla respiratoria o al facilitar la invasión de gérmenes al neonato por alterar procesos inmunológicos. Uno de cada 12 niños con PC tenía alguno de los 10 tipos de EIM encontrados. Es posible que muchas de las situaciones descritas como causales de PC (infecciones, hipoxia perinatal, dismetabolopatías, etc.) no sean más que consecuencias de la cadena de eventos iniciada por un daño primario en las estructuras celulares antes mencionadas.
CONCLUSIONES
Se tipificaron los errores innatos
metabólicos que, en número de 174, y con diez formas clínicas distintas, se
evidenciaron en pacientes con PC atendidos en un centro público
de Caracas, especializado en PC. Para diagnosticar hay que pensar y, en base a
ello, buscar; tomar en cuenta los «signos de alarma» descritos por edades, si
hay datos positivos: se recomienda aplicar la pesquisa completa. Instaurar
rápidamente el tratamiento de reemplazo y la dieta apropiada pues son infantes que pueden tener otras
patologías, ingresarlos en programas de Intervención Temprana y, más adelante, Integración
Educativa. Por las circunstancias
catastróficas de Venezuela en los últimos años, es posible que la casuística
sea varias veces mayor, el descarte neonatal es relativamente fácil efectuarlo
y dejó de hacerse en forma completa en los centros asistenciales públicos
hace casi dos decenios, y el segmento que sí se hacía, desapareció hace una
decena de años; el tratamiento es relativamente sencillo pero está fuera del
alcance del estrato social bajo que es al cual están insertos todos los casos
registrados en el estudio. De
no instaurarse la terapia requerida (rehabilitación, dieta y terapias de
reemplazo, luego, atención educativa según capacidades), se producen o acentúan
las secuelas, las cuales no revierten.
Agradecimiento
Agradecemos a las familias de los trabajadores del Centro de Parálisis Cerebral y de los
más de siete mil infantes que han asistido allí en los últimos 32 años, y sobre todo, las de los 2.000 infantes con parálisis
cerebral aquí revisados.
REFERENCIAS
1. Barkudah E, Glader L. Cerebral palsy: Epidemiology, etiology and prevention. https://www.uptodate.com/contents/search. Accessed July 10, 2019.
2. Ali A, Yalçın R, Ünlüer-Gümüştaş A. Cranial MR characteristics of Cerebral Palsy cases and correlation of findings with clinical results. Turk J Pediatr. 2019; 61(4): 525-537.
3. Stadskleiv K. Cognitive functioning in children with cerebral palsy. Dev Med Child Neurol. 2020 Mar; 62(3): 283-289.
4. Lemeshko V. Mitocondrias normales. El papel de la membrana mitocondrial externa en el control del metabolismo energético celular. Rev Acad Colomb Cienc Ex Fis Nat Enero-marzo 2018; 42(162): 6-21.
5. Rebecca V, Nicastri M, McLaughlin N, Fennelly C, McAfee
Q, Roughe A, et al. A Unified Approach to Targeting the Lysosome's Degradative and Growth Signaling Roles. Cancer Discov 2017; 7(11): 1266-1283. doi: 10.1158/2159-8290.CD-17-0741. Epub 2017 Sep 12
6. Garcia G. Los peroxisomas y su importancia biomédica. Rev Medic Sanitas 2019; 12 (2): 30-40.
7. D’Ortencio A, Navigante A. Disfunción mitocondrial y enfermedades cardiovasculares. Insuf Card 2016; 11 (4): 201-214.
8. Rua M. Enfermedades metabólicas lisosomales. Manifestaciones osteoarticulares. Protoc Diagn Ter Pediatr 2014; (1): 231-239.
9. Coutinho M. Molecular, biochemical and functional study in genes determining missorting of lysosomal proteins. ProQuest LLC, Ann Arbor, MI. (2017). P. 1-3.
10. Rosewich H, Waterham H, Tien B, Ohlenbusch A, Gärtner J. Clinical utility gene card for Zellweger syndrome Spectrum. European Journal of Human Genetics. 2015; 23. doi:10.1038/ejhg.2014.250; published online 19 November 2014.
11. Cabello J, Giugliani R. Errores innatos del metabolismo. Rev Med Clin Las Condes 2015; 26(4): 483-486.
12. López L, Gutiérrez L. Tratamiento de las enfermedades lisosomales en la población pediátrica. Anals Ped Contin 2013; 11(3): 159-161.
13. Mattman A, Potter M. Approach to the interpretation of unexpected laboratory results, arising in the care of patients with inborn errors of metabolism (IEM). Rev Endocr Metab Disord 2018 Mar; 19(1):5-12.
14. Sanjurjo P, Baldellou A, Aldámiz K, Montejo M, García M. Los errores congénitos del metabolismo como enfermedades raras con un planteamiento global específico. Anales Sis San Navarra 2008; 31(Suppl 2): 55-73.
15. Kleinsteuber K, Avaria M, Varela X. Parálisis cerebral. Rev Ped Electrón 2014; 11(2): 54-70.
16. Cabello J, Giugliani R. Errores innatos del metabolismo. Rev Med Clin Las Condes. 2015/07/01 doi10.1016/j.rmclc.2015.06.022
17. Rejingoud DJ. Flux analysis of inborn errors of metabolism. J Inherit Metabolism Dis 2018; 41(3): 309-28.
18. Campistol J. Orientación diagnóstica de los errores congénitos del metabolismo a partir de la neuroimagen. Pediatr Panamá 2017; 46 (2): 87-92.
19. Ruiz Pons M, Sánchez-Valverde F, Dalmau Serra J, Gómez L. Tratamiento nutricional de los errores innatos del metabolismo. 2a ed. Madrid Drug Farma 2017. Pp. 22-29.
20. Forero E, Echeverri O, Espinosa E, Guevara J, Barrera L. Acidemia glutárica tipo 1: presentación de un caso y revisión de la literatura. Iatreia. 2015 Abr-Jun;28(2):193-197. DOI 10.17533/udea.iatreia.v28n2a09.
21. Dominguez C, Martin M. Miopatías metabólicas, mitocondriales y tóxicas. Medicine 2019; 12(76): 4497-4506.
22. Pallarés M, Díaz M, Cervino J, Ripoll E, Martínez M. Acidosis láctica en pediatría. Quím Clín 2002; 21 (4): 280-284.
23. Brown G. Pyruvate dehydrogenase deficiency and the brain. Dev Med Child Neurol 2012; 54: 395-396.
24. Manoli I, Sloan J, Venditti C. Isolated Methylmalonic Acidemia. Gene Reviews. Dec 1, 2016; https://www.ncbi.nlm.nih.gov/books/NBK1231/. Accessed 1/21/2020.
25. Mora L, Suárez F. Aproximación diagnóstica de los errores innatos del metabolismo en la unidad de cuidado intensivo neonatal: acidosis metabólica, hiperamonemia e hipoglicemia. Revista Biosalud 2017; 16(2): 83-95 DOI: 10.17151/biosa.2017.16.2.8
26. Kontoangelos K, Lazaratou H, Economou M, Dikeos D, Papageorgiou C. Acute Psychotic Syndrome in a Male Adolescent with Succinic Semialdehyde Dehydrogenase Deficiency. Psychiatry Investig. 2019 Feb; 16(2): 172–173.
27. Mato S, Queiro V, Merino A, López M. Cribado neonatal del déficit de biotinidasa. Red Española de Agencias de Evaluación de Tecnologías y Prestaciones del SNS. Agencia de Evaluación de Tecnologías Sanitarias de Galicia; 2014.
28. Martins J, Teixeira M, Cardoso M, Lima P, Briones C. Galactosemia: genotipo y fenotipo de siete pacientes. Rev Neurol 2004; 38(12):1132-1135.
29. Spencer L, Bubner T, Bain E, Middleton P. Screening and subsequent management for thyroid dysfunction pre‐pregnancy and during pregnancy for improving maternal and infant health. Cochrane Database of Systematic Reviews 2015; 9: CD011263. DOI: 10.1002/14651858.CD011263.pub2.
30. Sanchez A, Martínez L, Arteaga G, Torres R,
Marroquín A, Abrego V, et al. Secuelas neurológicas en tres pacientes con fenilcetonuria clásica diagnosticada tardíamente. Bol. Med. Hosp. Infant. Mex. [online]. 2008; 65(3): 191-195.
Información adicional
Conflictos de interés: Los autores declaran no tener conflictos de intereses
Cómo citar: Rodríguez JM, Sánchez J. Errores innatos metabólicos en Parálisis Cerebral. Estudio en 174 infantes. Rev Digit Postgrado. 2020; 9(2): e205. doi: 10.37910/RDP.2020.9.2.e205.




