

Artículos de revisión
Aspectos clínicos, etiológicos y terapéuticos de la disqueratosis congénita
Clinical, etiological and therapeutic aspects of Dyskeratosis Congenita
 Francisco Cammarata-Scalisi francocammarata19@gmail.com
Francisco Cammarata-Scalisi francocammarata19@gmail.com
Revista Peruana de Investigación en Salud
Universidad Nacional Hermilio Valdizán, Perú
ISSN: 2616-6097
ISSN-e: 2616-6097
Periodicidad: Semestral
vol. 4, núm. 2, 2020
Recepción: 07 Enero 2020
Aprobación: 18 Enero 2020

Resumen: La disqueratosis congénita corresponde la primera entidad genética descrita entre las telomeropatías, cuya forma clásica se caracteriza por presentar la tríada mucocutánea de pigmentación reticulada de encaje en piel, distrofia ungueal y leucoplasia oral. Puede cursar además con insuficiencia de la médula ósea, tumores hematológicos y sólidos que corresponde las complicaciones más graves. Además de inmunodeficiencias, alteraciones dentales, pulmonares y hepáticas y otros aspectos considerados menores. Presenta a su vez una variada heterogeneidad genética de locus, con al menos 14 genes implicados en el acortamiento de los telómeros, asociados por lo tanto a la disqueratosis congénita o a fenotipos similares. Esta revisión discute además de las características clínicas, las diversas causas etiológicas, evolución, opciones terapéuticas disponibles y diagnóstico diferenciales de esta entidad con el objeto de brindar una evaluación médica interdisciplinaria e individualizada que incluya un adecuado asesoramiento genético.
Palabras clave: disqueratosis congénita, telomeropatías, clínica, etiología, tratamiento.
Abstract: The Dyskeratosis congenita corresponds to the first genetic entity described among telomeropathies, whose classic form is characterized by presenting the mucocutaneous triad of reticulated skin-lace pigmentation, nail dystrophy and oral leukoplakia. It can also occur with bone marrow failure, hematological and solid tumors, corresponding to the most serious complications. In addition to immunodeficiencies, dental, lung and liver disorders and other aspects considered minor. In turn, it presents varied genetic locus heterogeneity, with at least 14 genes involved in the telomere´s shortening, therefore associated with dyskeratosis congenita or similar phenotypes. This review discusses in addition to the clinical characteristics, the various etiological causes, evolution, available therapeutic options, and the differential diagnostic of this entity in order to provide an interdisciplinary and individualized medical evaluation that includes adequate genetic counseling.
Keywords: dyskeratosis congenita, telomeropathies, clinic, etiology, treatment.
Introducción
Los trastornos en la biología de los telómeros o telomeropatías, representan un grupo de entidades genéticas, en que la disqueratosis congénita (DC), fue la primera en describirse (1), cuya forma clásica se caracteriza clínicamente por presentar la tríada mucocutánea diagnóstica, de pigmentación reticulada de encaje en piel (1,2), que afecta principalmente el área del cuello y el tórax anterosuperior (2), distrofia ungueal y leucoplasia oral (1,2,3,4,5,6,7,8,9,10) (Fig. 1).
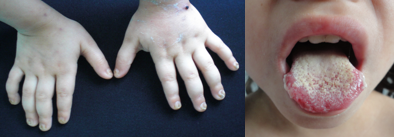
Las manifestaciones dermatológicas típicas ocurren en los primeros años de vida, sin embargo, la presentación puede ser heterogénea (8,9). La ya comentada distrofia ungueal afecta primero a las uñas de las manos, comienza con surcos y divisiones longitudinales, el cual progresa dando como resultado uñas rudimentarias, pequeñas o ausentes. Por su parte, la leucoplasia afecta la mucosa bucal, la lengua y la orofaringe y el tratamiento es sintomático. Aproximadamente, 30% presentan transformación maligna a carcinoma de células escamosas (4), por lo que requieren un seguimiento frecuente con realización de biopsia temprana en áreas de sospecha (4,8).
Posteriormente, se demostró que se asociaba a insuficiencia de la médula ósea (1,2,3,4,5), con citopenia de uno o más linajes de células hematopoyéticas y correspondió la complicación más grave (1). Puede afectar hasta 80-90% de los casos a la edad de 30 años y sus secuelas representan más de 70% de las muertes en pacientes con esta entidad (8). La presencia de la tríada mucocutánea puede ayudar a diferenciar la DC de otros tipos de insuficiencia de médula ósea (10). Además cursa con inmunodeficiencias, predisposición a presentar caries dental, hipodoncia, sangrado, recesión y pérdida ósea que simulan a la periodontitis juvenil, taurodontismo, inflamación gingival, y pigmentación intraoral parda (1,2,4,6,8,10). La evidencia de múltiples dientes permanentes con disminución de la relación raíz/corona pueden sugerir el diagnóstico de DC (4). Entre otros hallazgos clínicos se encuentran fibrosis pulmonar, e insuficiencia o fibrosis hepática (1,2,3,5,7,10). Presentan además un riesgo de 50 veces mayor de la población general de desarrollar tumores hematológicos y sólidos, incluidos el síndrome mielodisplásico, la leucemia mieloide aguda, el linfoma no Hodgkin, el carcinoma de células escamosas de cabeza y cuello, el cáncer de esófago, anogenital, y el carcinoma de células basales, como complicaciones graves (1,2,4,5,8,10).
El carcinoma de células escamosas de cabeza y cuello, es el tumor sólido más frecuente en pacientes con DC, con una edad media de inicio de 32 años, en comparación de 67 años en la población general.
El reconocimiento incluye características menores adicionales como el retraso del crecimiento intrauterino, retraso en el desarrollo, microcefalia, signos de envejecimiento prematuro, encanecimiento precoz del cabello, sudoración excesiva, y talla baja (4,7,8,9,10). Entre las anomalías oculares se describen la estenosis del conducto nasolagrimal, epífora, blefaritis, pestañas escasas, ectropión, entropión y triquiasis (2,3,4,10). Por su parte, los cambios retinianos son poco frecuentes e incluyen hemorragias, infarto de la capa de la fibra nerviosa, arteriosclerosis, edema macular, fibrosis preretinal y atrofia óptica (4). Además se puede evidenciar miocardiopatía, enteropatía con malabsorción, estenosis esofágica y uretral, hipogonadismo, atrofia testicular, osteoporosis, y necrosis avascular de las articulaciones de hombros y caderas (2,3,4,7,10). Estas manifestaciones clínicas afectan a los pacientes de diferentes formas y con un porcentaje variable (4). La DC pasa a menudo desapercibida debido al inicio tardío de los hallazgos mucocutáneos y en algunos casos pueden no presentarse. El amplio espectro de presentación clínica y la falta de pruebas de laboratorios concluyentes a veces pueden hacer que el diagnóstico clínico sea un reto (1).
En la DC como otras enfermedades de los telómeros, los síntomas van asociados al grado de acortamiento telomérico, causado por mutación en los genes de la telomerasa. En los casos más graves y con acortamiento telomérico menor al percentil 1% de la población, la enfermedad aparece en los primeros 10 años de vida o incluso durante la etapa prenatal. En los casos de acortamiento telomérico menos severo, la enfermedad aparece a una edad entre 15 y 25 años, en estos casos está por debajo del percentil 10% de la población y puede aparecer como aplasia medular o fibrosis pulmonar y cuya probabilidad está asociada al gen mutado. El diagnóstico genético es esencial debido a la limitada eficacia de las opciones terapéuticas, y la anticipación genética común en la DC hace que el asesoramiento genético sea una prioridad (1).
Incidencia
La DC presenta una incidencia anual estimada de menos 1 en 1.000.000(4), fue descrita por primera vez por Zinsser en 1906, en pacientes masculinos con la tríada mucocutánea anteriormente comentada (3,4,6). Fue reconocida como entidad clínica por Engman en 1926 y Cole en 1930, y se conoce como el síndrome de Zinsser-Cole-Engman, el cual se discutirá posteriormente (4). En 1963, se documentó el primer caso en el sexo femenino (3).
Etiopatogenia y diagnóstico
Los telómeros, están constituidos por repeticiones de 6 nucleótidos en los extremos de los cromosomas (5). Es un complejo de nucleoproteínas esencial para la estabilidad cromosómica, y se acortan con cada división celular (5,7). La regulación de la longitud de los telómeros está implicada en el envejecimiento celular y la tumorigénesis (6). Por lo tanto, mutaciones en los genes claves de la maquinaria del mantenimiento de los telómeros, se encuentran relacionados con entidades como la DC, que pueden causar insuficiencia de la médula ósea y cáncer (6,7,9,10).
Este conocimiento permitió desarrollar una prueba diagnóstica a través de la citometría de flujo con la hibridación fluorescente in situ, en subconjuntos de leucocitos. El tamaño de los telómeros en los leucocitos menor al primer percentil para la edad, es sensible en más de 95% y específico en pacientes con DC (3,5,7). Además de intervenir en el diagnóstico, el estudio de los telómeros ha contribuido enormemente a descubrir las causas genéticas de la DC (3).
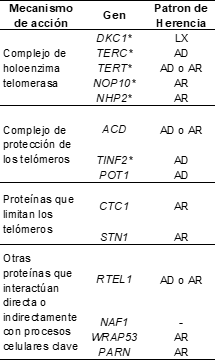
Se han identificado al menos 14 genes implicados en el acortamiento de los telómeros, asociados con la DC o fenotipos similares (Tabla 1), y representan entre 70-80% de los pacientes con esta entidad (3,5,7). Exhiben diversos patrones de herencia, entre ellos el ligado al cromosoma X recesivo (OMIM #305000)(1,3,4,5), debido a variantes patógenas en el gen disquerina 1 (DKC1), que codifica una pseudouridina sintasa, y cuyo gen se encuentra localizado en Xq28, entre los más comunes y clásicos de la DC, así como patrones de herencia autosómico dominante (OMIM #127550) y/o recesivo (OMIM #224230) (1,2,3,4,5,6,8,9). Sin embargo, entre 20-40% de los casos, las causas genéticas son indetectables (2,4).
La DC asociada al cromosoma X ocurre en el sexo masculino. Se manifiesta clínicamente entre los 5 a 12 años, puede presentar una edad de inicio variable, síntomas y severidad, incluso en individuos con la misma mutación. En este patrón de herencia, las mujeres pueden exhibir características clínicas menos graves a edades más avanzadas (4). El gen DKC1 presenta una secuencia muy conservada que se une a los ARN pequeños nucleolares, responsable de la biogénesis de los ribosomas, y forma parte del complejo de la telomerasa cuando se une al ARN de la telomerasa o TERC (2,9). Los estudios de Mitchell y Collins, fueron los primeros en mostrar una conexión entre los telómeros y la enfermedad humana a través de la función aberrante de la disquerina y el acortamiento de los telómeros (3). Por lo tanto, mutaciones en la línea germinal en los genes que intervienen en la biología de los telómeros dan como resultado acortamiento anormal de estas estructuras para la edad, que producen inestabilidad cromosómica y muerte celular progresiva (2).
Por lo tanto, es una enfermedad de mantenimiento defectuoso de los telómeros, que conlleva a un acortamiento prematuro, senescencia replicativa, agotamiento prematuro de las células madre y a fallo en diferentes tejidos, y se manifiesta principalmnte en tejidos mucocutáneos altamente proliferantes (1, 4, 10). Sin embargo, también se han identificado mutaciones en los componentes de la telomerasa y telómero en pacientes con anemia aplásica, fibrosis pulmonar y enfermedades hepáticas (4).
Adicionalmente, la correlación genotipofenotipo es compleja debido a varios factores, incluidos una variedad de mutaciones de genes hipomórficos subyacentes, anticipación de la enfermedad, así como efectos modificadores genéticos y ambientales (4).
Tratamiento
No hay terapia curativa para la DC y los pacientes generalmente mueren de forma prematura por insuficiencia de la médula ósea, debido a la deficiente capacidad de renovación de las células madres hematopoyéticas, por lo que una opción es el trasplante alogénico, el cual puede estar asociado a complicaciones. Además, el seguimiento es crucial ante la presencia de tumores, o infección grave por agentes oportunistas, entre las principales causas de muerte entre la segunda y tercera década de la vida (2,4,5,11).
El tratamiento del carcinoma de células escamosas de cabeza y cuello puede implicar cirugía, radiación y quimioterapia, a su vez se debe evitar la exposición a carcinógenos potenciales como la radiación ultravioleta, el alcohol, y el tabaco (8).
El uso de bajas dosis de retinoides sistémicos, ha evidenciado cierta mejoría en piel y uñas, pero los efectos secundarios y a largo plazo son inciertos (11). Por otra parte, se ha descrito que hasta un 70% de los pacientes con DC e insuficiencia en la médula ósea grave se pueden beneficiar de forma transitoria con la terapia con andrógenos o derivados de andrógenos, pero no hay curación de la enfermedad (8,11). No se conocen los mecanismos biológicos por los cuales estos compuestos tratan efectivamente la insuficiencia de la médula ósea. No obstante, se ha propuesto que los andrógenos pueden aumentar directamente la producción de eritropoyetina o actuar sobre el receptor de eritropoyetina para provocar una respuesta hematológica. Un número limitado de estudios sobre líneas celulares humanas y modelos de ratón con anemia aplásica sugieren que los andrógenos pueden aumentar la expresión de la telomerasa y la longitud de los telómeros (8).
Otras terapias exógenas pueden corregir el defecto de la telomerasa y mejorar el crecimiento celular, así como el uso de moduladores involucrados en el mantenimiento de los telómeros, se han sugerido como nuevos métodos terapéuticos (11). Entre ellos, la expresión de un péptido derivado de la disquerina, un elemento supresor genético 24.2 (GSE24.2, por sus siglas en inglés), el cual aumenta la actividad de la telomerasa, reduce los efectos patogénicos de las mutaciones Dkc1, disminuye el daño al ADN y el estrés oxidativo, por lo que sugiere un nuevo enfoque terapéutico. Igualmente, la expresión de GSE4, activa los promotores de c-myc y TERT, además aumento la expresión de c-myc, TERT y TERC (12,13,14).
Por otra parte, la activación de la telomerasa mediante el uso de vectores en terapia génica de virus adenoasociados que llevan el gen Tert de la telomerasa en modelos de ratones independientes de anemia aplásica debido a telómeros cortos, encontró que una dosis alta de este vector se dirige al compartimento de la médula ósea, incluidas las células madres hematopoyéticas, mejorando la supervivencia, produciendo un aumento significativo de la longitud de los telómeros en las células de sangre periférica y de la médula ósea, así como un mejor conteo sanguíneo. Estos hallazgos indican que la terapia génica con telomerasa representa una estrategia terapéutica novedosa para tratar la anemia aplásica provocada o asociada con telómeros cortos (9,15).
La integración de un equipo interdisciplinario es independiente en cada caso, pero general-mente incluirá Dermatología, Otorrinolaringología, Odontología, Cirugía Maxilofacial, Oncología, Ginecología(8), Genética Médica e incluir pruebas genéticas tempranas para realizar un oportuno asesoramiento genético (8,11).
Evolución de la DC
Durante el seguimiento de la enfermedad al presentarse la triada mucocutánea, suele aparecer la insuficiencia la médula ósea. Sin embargo, a veces la manifestación de la enfermedad es ambigua. La anemia aplásica ocurre en una edad media de presentación de 11 años, es macrocítica, con niveles elevados de hemoglobina fetal. Inicia con trombocitopenia y durante la evolución, se generaliza y se desarrolla insuficiencia grave de la médula ósea. La mortalidad prematura puede ocurrir hasta el 80% de los casos por infecciones oportunistas. Por otra parte, pueden progresar en diferentes formas con la aparición de mielodisplasia en uno o más linajes (4).
Existe un acortamiento excesivo del telómero que puede llevar a inestabilidad genómica. Los estudios de microscopía electrónica revelaron que las células en la DC tienen un núcleo embrionario inmaduro y tienen una predisposición a sufrir transformación maligna. Además, la barrera del epitelio es menos efectiva que en el epitelio normal, por lo que hay una mayor permeabilidad de sustancias nocivas y carcinógenas en las capas germina-es. Por lo que, se observa un aumento en la tasa de transformación maligna en las áreas leucoplásicas, siendo necesario su monitorización periódica. Además, presentan una incidencia acumulada del riesgo de malignidad de 40-50% a los 50 años, por ejemplo, pueden desarrollar linfoma de Hodg-kin, carcinoma laríngeo y bronquial, adenocarcinoma del tracto gastrointestinal, entre otros a nivel genitourinario y esquelético (4).
Variantes clínicas
El síndrome de Zinsser-Cole-Engman o Hoyeraal-Hreidarsson (OMIM #305000), es la variante más grave y se caracteriza por insuficiencia progresiva de la médula, retraso del crecimiento intrauterino, del desarrollo, microcefalia, hipoplasia cerebelosa, retraso mental, inmunodeficiencia progresiva, y la triada mucocutánea, que suele llevar a la muerte en la primera infancia (1, 4, 6, 8, 10, 11, 16). Se debe a mutaciones en los genes DKC1 y RTEL1, principalmente debido a una disminución de la actividad telomerasa. Se puede hacer el diagnóstico si tiene cuatro o más de estos hallazgos, o si tiene hipoplasia cerebelosa y características adicionales de la DC (4).
El síndrome de Revesz (OMIM #268130), fue descrito por primera vez en 1992, se debe a mutaciones en el gen TINF2, es otra variante infrecuente de la DC, la característica definitoria es la retinopatía exudativa bilateral, que se presenta en la mayoría de los casos en asociación con la calcificación intracraneal y las alteraciones más clásicas de la DC, como la insuficiencia de la médula ósea y las anomalías mucocutáneas; además, puede presentar retraso del crecimiento intrauterino, hipoplasia cerebelosa, y retraso del desarrollo (4,6,11,17).
El síndrome de Coats plus (OMIM #612199), es un trastorno infrecuente con patrón de herencia autosómico recesivo, debido a mutaciones sin sentido en el gen CTC1, caracterizado por microangiopatía cerebroretiniana, con calcificaciones intracraneales, osteopenia y sangrado gastrointestinal (4,18,19).
Diagnóstico diferencial
El síndrome de Naegeli-Franceschetti-Jadassohn (OMIM #161000), se diferencia por la falta de leucoplasia, insuficiencia de la médula ósea y un mayor riesgo de neoplasia maligna (11). No obstante, este y la dermatopatía pigmentosa reticular (OMIM #125595), alteraciones alélicas, pueden tener una hiperpigmentación reticulada similar (20,21).
Por su parte, la anemia de Fanconi (OMIM #227650), generalmente presenta anomalías pigmentarias difusas o uniformes y la pancitopenia cursa con un inicio más temprano en comparación con la DC (11,16). Presenta además anomalías oculares, renales y en extremidades (16). Así como otros síndromes de rotura y reorganización cromosómica como el síndrome de Bloom (OMIM #210900)16,22, síndrome de rotura de Nijmegen (OMIM #251260), síndrome de Seckel (OMIM #210600), y finalmente el síndrome de pseudo-TORCH, debido a las calcificaciones cerebrales (22).
Entre otras entidades se encuentran el síndrome de Rothmund-Thomson (OMIM #268400), y la epidermólisis ampollosa simple (OMIM #131900) los cuales presentan pigmentación moteada con poiquilodermia similar (18,20). Además, el síndrome de Kindler (OMIM #173650), y la poiquilodermia con neutropenia tipo Clericuzio (OMIM #604173) (16,23,24). En los síndromes de Bloom, Kindler y Rothmund-Thomson las lesiones en piel pueden ser similares a las observadas en la DC, pero son más sensibles al sol y difieren en las características asociadas (16). Por último, los pacientes con enfermedad de injerto contra el huésped, tienen poiquilodermia, cambios en la mucosa similares al liquen plano, y distrofia ungueal evidentes posterior al trasplante de médula ósea (16,20).
Conclusión
Una amplia gama de alteraciones genéticas dan lugar a un amplio espectro de manifestaciones clínicas con edad de inicio variable, por lo que el diagnóstico puede ser un desafío, en este trastorno de la biología de los telómeros. Por lo anteriormente dicho, la DC es un síndrome hereditario, clínico y genéticamente heterogéneo de insuficiencia de la médula ósea y prototipo de este tipo de trastorno. Es por ello que la evaluación médica debe ser interdisciplinaria e individualizada en la que se debe brindar las opciones terapéuticas disponibles y un oportuno asesoramiento genético basado en el diagnóstico genético y estudios de longitud telomérica.
Referencias
1.Trotta L, Norberg A, Taskinen M, Béziat V, Degerman S, Wartiovaara-Kautto U, et al. Diagnostics of rare disorders: wholeexome sequencing deciphering locus heterogeneity in telomere biology disorders. Orphanet J Rare Dis. 2018;13:139.
2.Ratnasamy V, Navaneethakrishnan S, Sirisena ND, Grüning NM, Brandau O, Thirunavukarasu K, et al. Dyskeratosis congenita with a novel genetic variant in the DKC1 gene: a case report. BMC Med Genet. 2018;19:85.
3.Savage SA. Beginning at the ends: telomeres and human disease. F1000Res. 2018;7. pii: F1000 Faculty Rev-524.
4.Fernández García MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med. 2014; 5: 157-67.
5.Khincha PP, Bertuch AA, Gadalla SM, Giri N, Alter BP, Savage SA. Similar telomere attrition rates in androgen-treated and un-reated patients with dyskeratosis congenita. Blood Adv. 2018;2:1243-9.
6.Olivieri C, Mondino A, Chinello M, Risso A, Finale E, Lanciotti M, et al. Clinical hete-rogeneity in a family with DKC1 mutation, dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome in first cousins. Pediatr Rep. 2017;9:7301.
7.Ungar RA, Giri N, Pao M, Khincha PP, Zhou W, Alter BP, et al. Complex phenotype of dyskeratosis congenita and mood dysregu-lation with novel homozygous RTEL1 and TPH1 variants. Am J Med Genet A. 2018;176:1432-7.
8.Bongiorno M, Rivard S, Hammer D, Kentosh J. Malignant transformation of oral leukoplakia in a patient with dyskeratosis congenita. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;124:e239-42.
9.Perona R, Machado-Pinilla R, Manguan C, Carrillo J. Telomerase deficiency and cancer susceptibility syndromes. Clin Transl Oncol. 2009;11:711-4.
10.Carrillo J, Martínez P, Solera J, Moratilla C, González A, Manguán-García C, et al. High resolution melting analysis for the identification of novel mutations in DKC1 and TERT genes in patients with dyskeratosis congeni-ta. Blood Cells Mol Dis. 2012;49:140-6.
11.Zhang J, Li M, Yao Z. Updated review of genetic reticulate pigmentary disorders. Br J Dermatol. 2017;177:945-59.
12.Iarriccio L, Manguán-García C, Pintado-Berninches L, Mancheño JM, Molina A, Perona R, et al. GSE4, a small dyskerin- and GSE24.2-related peptide, induces telomerase activity, cell proliferation and reduces DNA damage, oxidative stress and cell Senescence in dyskerin mutant cells. PLoS One. 2015 Nov 16;10(11):e0142980.
13.Manguan-Garcia C, Pintado-Berninches L, Carrillo J, Machado-Pinilla R, Sastre L, Pérez-Quilis C, et al. Expression of the genetic suppressor element 24.2 (GSE24.2) decreases DNA damage and oxidative stress in X-linked dyskeratosis congenita cells. PLoS One. 2014;9:e101424.
14.Machado-Pinilla R1, Sánchez-Pérez I, Murguía JR, Sastre L, Perona R. A dyskerin motif reactivates telomerase activity in X-linked dyskeratosis congenita and in telomerase-deficient human cells. Blood. 2008;111:2606-14.
15.Bär C, Povedano JM, Serrano R, Benitez-Buelga C, Popkes M, Formentini I, et al. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood. 2016; 127: 770-9.
16.Penmatsa C, Jampanapalli SR, Bezawada S, Birapu UK, Radharapu VK. Zinsser-Cole-Engman syndrome: A rare case report. J Clin Diagn Res. 2016;10:ZD07-9.
17.McElnea EM, van der Spek N, Smith O, Fitzsimon S, Patel CK, O'Marcaigh A. Revesz syndrome masquerading as bilateral cicatricial retinopathy of prematurity. J AAPOS. 2013;17:634-6.
18.Romaniello R, Arrigoni F, Citterio A, Tonelli A, Sforzini C, Rizzari C, et al. Cerebroretinal microangiopathy with calcifications and cysts associated with CTC1 and NDP mutations. J Child Neurol. 2013;28(12):1702-8.
19.Gu P, Jia S, Takasugi T, Smith E, Nandakumar J, Hendrickson E, et al. CTC1-STN1 coordinates G- and C-strand synthe-sis to regulate telomere length. Aging Cell. 2018:e12783.
20.Garofola C, Gross GP. Dyskeratosis Congenita. Source Stat Pearls. Treasure Island (FL): Stat Pearls Publishing; 2018.
21.Belligni EF, Dokal I, Hennekam RC. Prenatal and postnatal growth retardation, microcephaly, developmental delay, and pigmentation abnormalities: Naegeli syndrome, dyskeratosis congenita, poikiloderma Clericuzio type, or separate entity? Eur J Med Genet. 2011;54:231-5.
22.Dehmel M, Brenner S, Suttorp M, Hahn G, Schützle H, Dinger J, et al. Novel mutation in the DKC1 gene: neonatal Hoyeraal-Hreidarsson syndrome as a rare differential diagnosis in pontocerebellar hypoplasia, primary microcephaly, and progressive bone marrow failure. Neuropediatrics. 2016; 47: 182-6.
23.Anwar MI, Rashid A, Ghafoor R, Tahir M, Rao SU, Mir F. Kindler's syndrome: a report of five cases in a family. J Coll Physicians Surg Pak. 2014;24:763-5.
24.Mostefai R, Morice-Picard F, Boralevi F, Sautarel M, Lacombe D, Stasia MJ, et al. Poikiloderma with neutropenia, Clericuzio type, in a family from Morocco. Am J Med Genet A. 2008;146A:2762-9.
Información adicional
Revista Peruana de Investigación en Salud: ISSN: 2616 - 6097
Enlace alternativo
http://revistas.unheval.edu.pe/index.php/repis/article/view/606 (html)

